Разстройства на обмяната на медта МКБ E83.0
Към разстройствата в обмяната на медта се отнасят болестта на Menkes и болестта на Wilson. Те предствавляват рядко срещани заболявания.
СИНДРОМ НА МЕНКЕС
› Диагноза
› Лечение
› Прогноза
Въведение
Болест (синдром) на Менкес (Menkes disease), известна още и като болест на къдравите кости и болест на стоманените коси, е Х-свързано рецесивно невродегенеративно заболяване. Заболяването е причинено от мутации в гени, кодиращи протеина за транспортиране на мед ATP7A, при което е налице нарушен транспортът на мед, което води до дефицит на мед в тялото.
Както всички X-свързани рецесивни състояния, болестта на Menkes е по-често срещана при мъжете, отколкото при жените.
За първи път заболяването е описано от John Hans Menkes през 1962 година, а през 1972 г. Danks за първи път отбелязва, че метаболизмът на медта е нарушен.
Момиче с фенотип на болестта на Менкес и X-автозомна хромозомна транслокация е описано през 1987 година, което води до идентифицирането на локуса на Х хромозомата през 1993 г. По-леките варианти на болестта на Менкес, включително синдром на тилния рог също са описани.
Характерните находки включват чуплива коса, забавяне на растежа и влошена функция на нервната система.
Етиология
Болестта на Менкес се развива в резултат на мутации на гена ATP7A, намиращ се в хромозома Xq13.3.
Това е X-свързано рецесивно заболяване, като около 30% от случаите се дължат на нови мутации, а 70% са унаследени, почти винаги от майката. Въпреки че заболяването е по-често при мъжете, жените могат да бъдат носители на болестта.
В резултат на мутацията медта се разпределя слабо в клетките на тялото, като се натрупва в някои тъкани (тънки черва и бъбреците), докато мозъкът и другите тъкани имат необичайно ниски нива. Намаленото снабдяване с мед може да намали активността на множество мед съдържащи ензими, които са необходими за структурата и функцията на костите, кожата, косата, кръвоносните съдове и нервната система - например лизил оксидаза.
Много от генните дефекти са делеции, които могат да бъдат открити чрез саутърн блот (Southern blot), но също така се съобщава за малки дупликации, нонсенс (nonsense) мутации и миссенс мутации.
Леките варианти на заболяването при хората обикновено се причиняват от сплайсинг дефекти.
Епидемиология
Честотата на това заболяване е от 1 на 50 000 до 1 на 250 000 раждания, като една трета от случаите са резултат от нови мутации. Не е известна връзка с други наследствени характеристики или с етнически произход.
Тъй като болестта на Менкес е Х-свързано рецесивно заболяване, обикновено засяга момчета чрез незасегнати жени-носителки. Заболяването може да се дължи на мозаицизъм на зародишната линия.
Съобщава се за няколко засегнати жени с X-автозомни транслокации, X0/XX мозаицизъм или неблагоприятна лионизация.
Началото на класическата форма е в ранна детска възраст. По-леките варианти започват в детството или ранната зряла възраст.
Патофизиология
Медта е елемент, който взема участие в много основни ензимни системи, включително:
- цитохром С оксидаза
- супероксид дисмутаза
- лизил оксидаза
- тирозиназа
- аскорбинова киселина оксидаза
- церулоплазмин
- допамин бета хидроксилаза
Счита се, че дефицитът или нарушената функция на тези ензимни системи са причина за клиничните прояви на синдрома на Менкес.
Гените на болестта на Menkes и Wilson имат 55% аминокиселинна идентичност, като АТФ-азите при болестта на Менкес и болестта на Уилсън имах сходни биохимични механизми, но се различават по тъканно-специфична експресия. Генът на болестта на Wilson се експресира предимно в черния дроб, докато генът на болестта на Menkes не се експресира в черния дроб, като предоминантните (основните) места на експресия на гена на Menkes са плацентата, стомашно-чревния тракт и кръвно-мозъчната бариера. Протеинът на Менкес също така се среща е в пигментните епителни клетки на ретината.
При болестта на Менкес, транспортът на приетата с храната мед от чревните клетки е нарушен, което води до ниски серумни нива на мед. Нарушеният транспорт на мед в други клетки води до прекомерно натрупване на мед в дуоденалните клетки, бъбреците, панкреаса, скелетните мускули и плацентата.
Нарушената активност на цитохром С оксидазата вероятно е причина за повечето от неврологичните симптоми, подобни на тези при пациенти с болест на Leigh (подостра некротизираща енцефаломиелопатия), които имат намалена или липсваща активност на цитохром С оксидазата и подобни невропатологични промени.
Намалената активност на лизилоксидазата е причина за слабост на съединителната тъкан и съдовите аномалии при болестта на Menkes, тъй като лизилоксидазата дезаминира лизин и хидроксилизин в процеса на синтез на колаген.
Дефицитът на тирозиназа (участваща в биосинтезата на меланин) най-вероятно е причината за хипопигментацията на косата и кожата, наблюдавана при болестта на Menkes.
Клинична картина
Засегнатите бебета могат да се родят преждевременно. Признаците и симптомите се появяват през ранна детска възраст, обикновено след период от 2 до 3 месеца след раждането, като са налице:
- забавено развитие
- мускулна хипотония
- епилепсия, разделена на 3 периода - 1 - ранен стадий (средна възраст 3 месеца); 2 - междинен стадий (средна възраст 10 месеца); късни прояви (средна възраст 25 месеца)
При пациентите се установява още хипотермия, отпуснати черти на лицето, гърчове и разширяване на метафизите (участъци от дългите кости). Косата има странен вид: къдрава, безцветна или сребриста и чуплива.
Наличието на остеопороза може да доведе до фрактури (счупвания).
Хората с по-леки варианти могат да имат минимални неврологични симптоми с нормален интелект или само леки интелектуални затруднения и автономна дисфункция.
Синдромът на тилния рог е лека форма на синдрома на Menkes, която започва в ранна до средна детска възраст.
Пациентите със синдром на тилния рог са засегнати предимно от промени в съединителната тъкан и костите, включително хипереластична и лесно образуваща синини кожа, халтави стави, хернии, дивертикули на пикочния мехур, груба коса и множество скелетни аномалии, включително калциеви отлагания по тилната (окципиталната) кост - "рога".
Рогата може да не присъстват в ранна детска възраст. Тези пациенти също могат да имат леки интелектуални затруднения и автономна дисфункция. Серумните нива на мед и церулоплазмин са ниски, но не в същата степен, както при болестта на Уилсън.
Окципиталните рога могат да присъстват и при пациенти с класическата форма на болестта на Menkes и са забелязани при пациенти на 2-годишна възраст.
Друг клиничен вариант, означаван като лека форма на болестта на Menkes, се характеризира с атаксия и леки интелектуални затруднения.
Диагноза
За диагнозата на болестта на Менкел се прилагат различни методи, включително:
Лабораторни изследвания
Към лабораторните изследвания, спомагащи за диагностициране на този синдром, спадат:
Изследване на нивата на мед и церулоплазмин
Нивата на мед и церулоплазмин могат да бъдат нормални при по-леките варианти, както и в неонаталния период. Общото съдържание на мед в тялото може да бъде нормално при кърмачето до 2 седмици след раждането или до по-късен етап.
Нивата на церулоплазмин са 6–12 mg/dL първоначално и едва по-късно се считат за патологично ниски. Нормално доносените новородени също имат по-ниски нива на мед в серума, а при недоносени бебета са още по-ниски нива.
Диагностичните находки включват:
- серумно ниво на мед под 70 mg/dL (референтни граници 80–160)
- серумно ниво на церулоплазмин под 20 mg/dL (референтни граници 20-60)
Плазмени катехоламини
При изследването на катехоламини може да се установи понижено ниво на норепинефрин, а също така може да се наблюдава повишено съотношение на хидроксифенилаланин (DOPA) и дихидроксифенилгликол (DHPG) поради намалена активност на допамин бета-хидроксилазата, като по-високите стойности отразяват по-тежко заболяване.
Диагностичните находки включват:
- стойности над 5 в серума (норма 1,7–3,3)
- стойности над 1 в цереброспиналната течност (норма 0,3–0,7)
Асимптоматични бебета с риск от болестта на Менкес могат да бъдат разделени на засегнати и незасегнати въз основа на плазмените неврохимични профили.
Други отклонения
Други лабораторни находки могат да включват:
- повишени нива на мед в червата и бъбреците
- намалени нива на мед в черния дроб
- хипогликемия
Дезоксипиридинолин (D-Pyr): D-Pyr се синтезира от лизил оксидаза, която е дефектна при болестта на Menkes. Нивата на D-Pyr в урината може да останат ниски въпреки лечението.
Образни изследвания
Компютърна томография (КТ) и ядрено-магнитен резонанс (ЯМР)
Данните от КТ и ЯМР могат да включват:
- демиелинизация на бялото мозъчно вещество
- други патологични промени на бялото вещество могат да бъдат симетрични
- деформирани кръвоносни съдове
- атрофични изменения
- субдурални хематоми и изливи
Рентгеново изследване
Рентгенография на черепа и тялото се прави с цел установяване аномалии в развитието на костите.
ЯМР спектроскопия
Спектроскопията с протонен магнитен резонанс показва повишен лактат и намалено съотношение N-ацетил аспартат (NAA):общ креатин (tCr).
Електроенцефалография (ЕЕГ)
Данните от ЕЕГ в ранните етапи на заболяването включват:
- иктална ЕЕГ находка - Протичане на бавни пикови вълни и бавни вълни в задните области
- интериктална ЕЕГ находка - мултифокални и полиморфни бавни вълни или смесени бавни пикови вълни и бавни вълни
Констатациите в късен етап могат да включват:
- интериктална ЕЕГ находка - мултифокална активност с висока амплитуда, смесена с неправилни бавни вълни
Хистологично изследване
Аутопсия на мозъка
Данните от аутопсия на мозъка могат да включват:
- дифузна атрофия
- фокална дегенерация на неврони
- невронална загуба в малкия мозък, засягаща клетките на Purkinje
- анормална дендритна арборизация (така наречената "плачеща върба") и перисоматични процеси
- фокално аксонално увреждане
- увеличен брой и размер на митохондриите
- изразено намаляване на вътрешните гранулирани клетки
- еозинофилни сфероидни тела
Биопсия на кожата
Промените включват наличието на тънка нишки от аморфен еластин, свързани с множество микрофибрили
Биопсия на мускул
Промените включват натрупване на гликоген и преобладаване на влакна тип 2
Генетично изследване
Тъй като заболяването е наследствено, може да се извърши генетично изследване на майката за търсене на мутация в гена ATP7A.
Лечение
За болестта на Менкес няма лечение.
Ранното прилагане на инжекции с медни добавки (ацетат или глицинат) може да бъде от известна полза.
Пероралното (през устата) лечение с медни соли като сулфат, ацетат или хлорид не променя серумните нива на мед и церулоплазмин. Парентерално (венозно) приложена мед индуцира синтеза на апоцерулоплазмин и WND гена, което води до повишаване на серумните нива на мед и церулоплазмин, но церебралните нива на мед не се променят и не настъпва клинично подобрение.
Новородени, както и фетуси (фетус = плод), третирани in utero с меден хистидин, могат да избегнат неврологичните симптоми. Когато обаче неврологичните симптоми веднъж са налице, са по-малко податливи на лечение.
Като цяло, лечението е симптоматично и поддържащо. Терапията за облекчаване на някои от симптомите включват болкоуспокояващи, лекарства против гърчове, сонда за хранене, когато е необходимо, както и физическа и трудова терапия.
Целите на лекарствената терапия са облекчаване на симптомите и предотвратяване на усложнения.
Колкото по-рано се започне с терапията, толкова по-добра е прогнозата.
В семейства с предишно засегнато дете може да се направи генетична консултация и пренатално изследване при бъдещи бременности.
Бебетата, които носят мутацията, трябва да бъдат идентифицирани и лекувани много рано в живота, преди да настъпи необратима невродегенерация.
Прогноза
Повечето нелекувани пациенти с класическата форма на болестта на Menkes умират до 3-годишна възраст.
БОЛЕСТ НА УИЛСЪН (Wilson)
› История
› Диагноза
› Лечение
› Прогноза
Болестта на Уилсън (Wilson disease) е рядко автозомно-рецесивно наследствено заболяване на метаболизма на мед в организма, което се характеризира с прекомерно отлагане на мед в черния дроб, мозъка и други тъкани. Заболяването често е фатално, ако не бъде разпознато и лекувано навреме.
Болестта на Wilson се причинява от мутация в гена на протеина на болестта на Wilson (ATP7B). Този протеин транспортира излишната мед в жлъчката, с която се екскретира от тялото.
История
Заболяването носи името на британския лекар Samuel Alexander Kinnier Wilson (1878–1937) - невролог, който описва състоянието, включително патологичните промени в мозъка и черния дроб, през 1912 г. Работата на Уилсън е предшествана от и се основава на доклади на немския невролог Karl Westphal (през 1883 година), който я нарича "псевдосклероза", на британския невролог William Gowers (през 1888 година), на финландския невропатолог Ernst Alexander Homén (през 1889–1892 година), който отбелязва наследствения характер на заболяването, както и на доклади от Adolph Strümpell (през 1898 година), който установява чернодробна цироза. Невропатологът John Nathaniel Cumings прави връзка с натрупването на мед както в черния дроб, така и в мозъка през 1948 година. Появата на хемолиза е отбелязана през 1967 година.
През 1951 г. Cumings и неврологът от Нова Зеландия Derek Denny-Brown, работещи в Съединените щати, едновременно съобщават за първото ефективно лечение, използващо метален хелатор.
Първият ефективен перорален хелатиращ агент - пенициламин - е открит през 1956 г. от британския невролог John Walshe.
Епидемиология
В световен мащаб честотата на болестта на Уилсън е 10-30 милиона случая, а хетерозиготното носителство е 1 случай на 100 души, като честотата на генетичните мутации варира от 0,3%-0,7%. Повишената честота в някои страни се дължи на високите нива на кръвно родство. Фулминантното (свръхостро) представяне на болестта на Wilson е по-често при жените, отколкото при мъжете.
Прояви, свързани с възрастта
Проучване на хора с болест на Wilson показва, че при пациентите, при които заболяването се проявява се по-рано, се установяват предимно чернодробни симптоми, докато тези, с по-късно начало по-често имат неврологични симптоми.
По принцип горната възрастова граница за развитие на болестта на Wilson е 40 години, а долната възрастова граница е 5 години, въпреки че заболяването е установено при деца под 3-годишна възраст и при възрастни над 70 години.
Етиология
Приблизителното общо съдържание на мед в тялото е 50-100 mg, а средният дневен прием е 2-5 mg в зависимост от индивидуалния хранителен режим. Медта е важен компонент на няколко метаболитни ензима, включително лизил оксидаза, цитохром с оксидаза, супероксид дисмутаза и допамин бета-хидроксилаза.
Около 50%-75% от приетата с храната мед се абсорбира и след това се транспортира до хепатоцитите (чернодробните клетки), където се включва в съдържащи мед ензими и мед-свързващи протеини, включително церулоплазмин, серумна фероксидаза.
При болестта на Уилсън процесите на включване на мед в церулоплазмин и екскреция на излишната мед с жлъчката са нарушени.
Излишната мед в резултат на болестта на Уилсън стимулира образуването на свободни радикали, което води до окисляване на липиди и протеини. Първоначално излишната мед се натрупва в черния дроб, което води до увреждане на хепатоцитите, а когато нивата на мед в черния дроб се повишат, нейните нива се увеличава и в кръвообращението, което води до отлагане и в други органи.
Патофизиология
Медта е необходима на тялото за редица функции, предимно като кофактор за редица ензими като церулоплазмин, цитохром оксидаза, допамин β-хидроксилаза, супероксид дисмутаза и тирозиназа.
Медта навлиза в тялото през храносмилателния тракт. Транспортен протеин в клетките на тънките черва - меден мембранен транспортер 1 - пренася медта вътре в клетките, а излишната мед се ексретира (отделя) чрез жлъчката.
При болестта на Wilson медта се натрупва в чернодробната тъкан, като церулоплазмин все още се секретира, но във форма, в която липсва мед (наречена апо-церулоплазмин) и бързо се разгражда в кръвната циркулация.
Когато количеството мед в черния дроб превиши нивото на свързващите протеини, се стига до окислително увреждане, което води до хроничен активен хепатит, фиброза и цироза. Черният дроб също освобождава мед в кръвта, която не е свързана с церулоплазмин. Тази свободна мед се отлага във всички тъкани, но особено в бъбреците, очите и мозъка. В мозъка по-голямата част от медта се отлага в базалните ганглии - тези области участват в координацията на движенията и играят значителна роля в неврокогнитивните процеси като обработката на стимули и регулирането на настроението. Увреждането на тези области предизвиква невропсихични симптоми, наблюдавани при болестта на Уилсън.
Високото ниво на свободна (несвързана с церулоплазмин) мед има пряк ефект върху окисляването на хемоглобина, инхибирането на доставящи енергия ензими в червените кръвни клетки или директно увреждане на клетъчните мембрани, което води до хемолиза.
Клинична картина
Болестта на Wilson може клинично да се прояви по различен начин - от асимптоматично състояние до фулминантна чернодробна недостатъчност, хронично чернодробно заболяване със или без цироза, неврологични и психични прояви.
Чернодробно заболяване
За болест на Wilson трябва да се мисли при всяко необяснимо хронично чернодробно заболяване, особено при лица под 40 години. Състоянието може да се прояви и като остър хепатит. Чернодробната дисфункция се проявява при повече от половината пациенти. Трите основни модела на чернодробно засягане включват: (1) хроничен активен хепатит, (2) цироза и (3) фулминантна чернодробна недостатъчност. Най-честата първоначална проява е цироза.
Чернодробното заболяване може да се прояви с умора, жълтеница, повишена склонност към кървене, дезориентация (поради чернодробна енцефалопатия) и портална хипертония. Порталната хипертония води до варици на хранопровода, (разширени кръвоносни съдове в хранопровода, които могат да кървят животозастрашаващо), уголемяване на далака (спленомегалия) и натрупване на течност в коремната кухина (асцит).
При преглед може да се наблюдават признаци на хронично чернодробно заболяване като паякообразни ангиоми (малки разширени кръвоносни съдове).
Докато повечето хора с цироза имат повишен риск от хепатоцелуларен карцином, този риск е относително много нисък при болестта на Wilson.
Около 5% от всички хора се диагностицират едва когато развият фулминантна чернодробна недостатъчност, често съчетана с хемолитична анемия. Това води до аномалии в производството на протеини и метаболизма в черния дроб, което води до натрупване на остатъчни метаболити като амоняк в кръвта. Когато те достигнат мозъка, може да се развие чернодробна енцефалопатия.
Невропсихични симптоми
Приблизително 50% от пациентите с болест на Wilson имат неврологични или психични симптоми. Повечето пациенти, които имат невропсихични прояви, имат цироза. Най-честата неврологична характеристика е асиметричният тремор, който се среща при приблизително половината от индивидите с болестта на Wilson. Характерът на тремора е променлив и може да бъде предимно в покой, постурален или кинетичен. Пръстените на Kayser-Fleischer се наблюдават при поне 98% от пациентите с неврологична болест на Wilson, които не са получавали хелатотерапия.
Честите ранни симптоми включват затруднения в говора, прекомерно слюноотделяне, атаксия, масковидно лице, затруднено движение на ръцете и промени в личността.
Късните прояви (сега рядкост поради по-ранна диагностика и лечение) включват дистония, спастичност, гърчове, ригидност (повишен мускулен тонус) и флексионни контрактури.
Психичните прояви включват емоционална лабилност, импулсивност и самонараняващо се поведение. Тези прояви се установяват при 10%-20% от пациентите.
Мускулно-скелетни симптоми
Засягането на скелета е често срещана характеристика на болестта на Wilson, като повече от половината пациенти проявяват остеопения (намаляване на костната плътност) при конвенционално радиологично изследване.
Артропатията при болестта на Wilson е дегенеративен процес, който наподобява преждевременен остеоартрит. Симптоматичното ставно заболяване, което се среща при 20%-50% от пациентите, обикновено възниква в края на хода на заболяването, често след 20-годишна възраст. Артропатията обикновено включва гръбначния стълб и големите стави, като коленни, бедрени и киткови.
Скелетните аномалии при пациенти с болест на Wilson включват също така остеопороза, остеомалация, рахит, спонтанни фрактури (счупвания) и полиартрит.
Хематологични симптоми
Хемолитичната анемия е рядко (10%-15%) усложнение на заболяването. Остра интраваскуларна (вътресъдова) хемолиза най-често възниква в резултат на оксидативно увреждане на еритроцитите от по-високата концентрация на мед.
При всеки пациент, при който настъпва остра чернодробна недостатъчност с интраваскуларна хемолиза, умерено повишение на серумните аминотрансферази и ниска серумна алкална фосфатаза или съотношение на алкалната фосфатаза към билирубин по-малко от 2, трябва да се подозира за болест на Wilson.
Бъбречни симптоми
Генът на болестта на Wilson се експресира в бъбречната тъкан, в резултат на което всички бъбречни прояви могат да бъдат първични или вторични на освобождаването на мед от черния дроб.
Клинично пациентите могат да приличат на тези със синдром на Fanconi.
Уролитиазата (камъни в бъбреците), открита при до 16% от пациентите с болестта на Wilson, може да бъде резултат от хиперкалциурия (повишено ниво на калций в урината).
Съобщава се за хематурия (кръв в урината) и нефрокалциноза (отлагане на калциеви соли в бъбреците), а протеинурия (наличие на белтък в урината) и пептидурия могат да възникнат преди лечението като част от болестния процес и след терапията като неблагоприятни ефекти на D-пенициламин.
Сърдени прояви
Кардиомиопатията е рядък проблем при болестта на Wilson, като може да доведе до сърдечна недостатъчност и ритъмни нарушения.
Хормонални нарушения
Хипопаратироидизъм, панхипопитуитаризъм ( липса на хормоните от предния дял на хипофизната жлеза), безплодие и повтарящи се спонтанни аборти са най-честите хормонални нарушения при това заболяване.
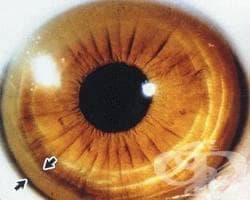
Офталмологични нарушения
Пръстените на Кайзер-Флайшер (Kayser-Fleischer) се образуват от отлагането на мед в десцеметовата мембрана на роговицата. Цветът може да варира от зеленикаво-златисто до кафяво. Когато са добре развити, пръстените могат да бъдат лесно видими с просто око или с офталмоскоп. Когато не се виждат с невъоръжено око, пръстените могат да бъдат идентифицирани чрез изследване с шлиц лампа или гониоскопия.
Пръстените на Kayser-Fleischer се наблюдават при до 90% от индивидите със симптоматична болест на Wilson и почти неизменно присъстват при тези с неврологични прояви.
Въпреки че пръстените на Kayser-Fleischer са полезен диагностичен знак, те вече не се считат за патогномонични за болестта на Wilson, освен ако не са придружени от неврологични прояви. Те могат да се наблюдават и при пациенти с хронични холестатични разстройства, като частична билиарна атрезия, първична билиарна цироза, първичен склерозиращ холангит и криптогенна цироза.
Диагноза
Наличието на пръстени на Kayser-Fleischer и нива на церулоплазмин под 20 mg/dL при пациент с неврологични признаци или симптоми предполагат диагноза болест на Wilson.
Повечето пациенти имат леко абнормени чернодробни функционални тестове, като повишени нива на АСАТ, АЛАТ и билирубин. Ако чернодробното увреждане е значително, албуминът може да бъде намален поради неспособност на увредените чернодробни клетки да произвеждат този протеин. По същата причина протромбиновото време може да бъде удължено, тъй като черният дроб не е в състояние да произвежда протеини, известни като фактори на кръвосъсирването. Нивата на алкалната фосфатаза са относително ниски при хора с остра чернодробна недостатъчност, свързана с болестта на Wilson.
Ако пациентът е асимптоматичен, установява се изолирано чернодробно заболяване, но липсват роговични пръстени (на Kayser-Fleischer), едновременното наличие на концентрация на мед в черния дроб над 250 mcg/g и ниско ниво на серумния церулоплазмин е достатъчно за насочване към тази на диагноза.
Златният стандарт за диагноза е чернодробна биопсия.
Генетична диагноза
Роднини от първа и втора степен на пациенти с потвърдена болест на Wilson трябва да бъдат изследвани за това състояние.
Образно изследване на корема
Компютърната томография (CT), ядрено-магнитен резонанс (MRI), ултразвуковото изследване и нуклеарномедицинските изследвания на черния дроб са недостатъчно информативни, с ниска специфичност и чувствителност чувствителни за болестта на Wilson.
Електрокардиография (ЕКГ)
Електрокардиографските аномалии в покой включват левокамерна или двукамерна хипертрофия, ранна реполяризация, депресия на ST сегмента, инверсия на Т-вълната и различни ритъмни нарушения.
Изследване с шлиц лампа
Очите на пациента се изследват с помощта на специална лампа, за да се търсят пръстени на Kayser-Fleischer (Кайзер-Флайшер), които са силно асоциирани с болестта на Wilson и са причинени от отлагане на мед в мембраната на Descemet на роговицата.
Серумен церулоплазмин
Серумните нива на церулоплазмин са ниски при новородени и постепенно се повишават през първите 2 години от живота. Приблизително 90% от всички пациенти с болест на Wilson имат нива на церулоплазмин под 20 mg/dL (референтен диапазон 20-40 mg/dL). Церулоплазминът е острофазов протеин и може да се повиши в отговор на чернодробно възпаление, бременност, употреба на естроген или инфекция.
Фалшиво ниски нива на церулоплазмин могат да се наблюдават при всяко състояние на протеинов дефицит, включително нефротичен синдром, малабсорбция, протеин-губеща ентеропатия и недохранване. Нивата на церулоплазмин също могат да бъдат намалени при 10%-20% от хетерозиготите на гена на болестта на Wilson, които не развиват болестта на Wilson и не се нуждаят от лечение.
Екскреция на мед в урината
Екскретираната мед в урината е повече от 100 mcg/ден (референтен диапазон < 40 mcg/ден) при повечето пациенти със симптоматична болест на Wilson. Нивата могат да бъдат повишени и при други холестатични чернодробни заболявания.
Чувствителността и специфичността на този тест не са оптимални за използване като скринингов тест, но може да бъде полезен за потвърждаване на диагнозата и за оценка на отговора на хелатотерапията.
Концентрация на мед в черния дроб
Този тест се счита за стандартен критерий за диагностика на болестта на Wilson. Чернодробна биопсия разкрива нива над 250 mcg/g дори при асимптоматични пациенти.
Нормалната концентрация на мед в черния дроб (референтен диапазон 15-55 mcg/g) ефективно изключва диагнозата нелекувана болест на Wilson.
Повишена концентрация на мед в черния дроб може да се открие при други хронични чернодробни (предимно холестатични) нарушения.
Компютърна томография на глава
Черепните лезии, наблюдавани при компютърна томография, обикновено са двустранни и се класифицират в две основни категории: (1) добре изразени, включващи базалните ганглии, особено путамена (2) по-големи области в базалните ганглии, таламуса или nucleus dentatus.
Ядрено-магнитен резонанс (ЯМР) на мозъка
ЯМР на мозъка е по-чувствителен метод от компютърната томография при откриване на ранни лезии на болестта на Wilson. ЯМР изследванията идентифицират фокални аномалии в бялото вещество, моста и дълбоките церебеларни ядра. Тези лезии, с размери 3-15 mm в диаметър, обикновено са двустранни и представляват загуба на клетки и глиоза.
PET сканиране
Сканирането с позитронно-емисионна томография (PET) разкрива значително намалена церебрална метаболитна скорост на утилизация на глюкоза в малкия мозък, мозъчната кора и таламуса.
Електронна микроскопия
Електронномикроскопското откриване на съдържащи мед хепатоцитни лизозоми е полезно при диагностицирането на ранните стадии на болестта на Уилсън, в допълнение към количественото определяне на чернодробната мед чрез абсорбционна спектрофотометрия.
Чернодробна биопсия
След като други изследвания са насочили към болестта на Wilson, окончателна диагноза се поставя чрез чернодробна биопсия. По този начин се оценява микроскопски степента на стеатоза и цироза, а хистохимията и количественото определяне на медта се използват за определяне на тежестта на натрупването на мед. Ниво от 250 μg мед на грам чернодробна тъкан потвърждава болестта на Wilson. Понякога се откриват по-ниски нива на мед, като в този случай комбинацията от резултатите от биопсията с всички други тестове все още може да доведе до окончателна диагноза на болестта на Wilson.
В по-ранните стадии на заболяването биопсията обикновено показва стеатоза, повишено ниво на гликоген в ядрата и области на некроза. При по-напреднало заболяване наблюдаваните промени са подобни на тези, наблюдавани при автоимунен хепатит, като инфилтрация от възпалителни клетки, частична некроза и фиброза. При напреднало заболяване цирозата е основната находка.
При остра чернодробна недостатъчност се наблюдава дегенерация на чернодробните клетки и колапс на архитектурата на чернодробната тъкан, обикновено на фона на циротични промени. Хистохимичните методи за откриване на мед са противоречиви и ненадеждни и взети сами по себе си се считат за недостатъчни за установяване на диагноза.
Лечение
Диета
Пациентите трябва да избягват да консумират храни с високо съдържание на мед - черен дроб, шоколад, ядки, гъби, бобови растения, сушени плодове, сусамово семе и сусамово масло, както и миди.
Питейната вода от нетипични източници (например вода от кладенци) трябва да се анализира за съдържание на мед и да се замени с пречистена вода.
Медикаментозно лечение
Основата на терапията за болестта на Wilson е фармакологично лечение с хелатиращи агенти като D-пенициламин и триентин. Други агенти включват натриев димеркаптосукцинат, димеркаптосукцинова киселина, цинк и тетратиомолибдат. Цинковите соли действат като индуктори на металотионеините, които намаляват резорбцията на мед и съответно на свободната плазмена мед.
Обикновено пенициламинът е първи избор на лечение. Той свързва медта (хелиране) и води до екскреция на медта в урината, като може да се извършва мониторинг на количеството мед в урината, за да се гарантира, че е приета достатъчно висока доза.
Пациентите с непоносимост към пенициламин могат вместо това да започнат с триентин хидрохлорид, който също има хелатни свойства.
След като всички резултати се нормализират, може да се използва цинк (обикновено под формата цинков ацетат) вместо хелатори за поддържане на стабилни нива на мед в тялото. Цинкът стимулира металотионеините - протеини в чревните клетки, които свързват медта и предотвратяват нейното усвояване и транспортиране до черния дроб. Цинковата терапия продължителна, освен ако симптомите не се появят отново или ако екскреция на мед в урината се увеличи.
В редки случаи, когато нито едно от пероралните лечения не е ефективно, особено при тежки неврологични заболявания, понякога е необходим димеркапрол. Този медикамент се прилага интрамускулно на всеки няколко седмици.
Хората, които са асимптоматични (например тези, диагностицирани чрез скрининг или само в резултат на необичайни резултати от тестове), обикновено се лекуват, тъй като натрупването на мед може да причини дългосрочни увреждания в бъдеще.
Бременност
Високите вътрематочни концентрации на мед може да са причина за високия процент на спонтанни аборти при пациенти с болест на Уилсън. Лечението с D-пенициламин не представлява голям риск за плода и трябва да продължи през цялата бременност.
Въпреки че бременността сама по себе си не изглежда да има вреден ефект върху хода на лекуваните пациенти, рискът от асцит или кървене от гастроезофагеални варици (разширени кръвоносни съдове, установени в хранопровода и стомаха) по време на бременност е повишен за всеки индивид с цироза, независимо от основната етиология.
Физикална и трудова терапия
Физиотерапията и трудотерапията са полезни при пациенти с неврологична форма на заболяването. Лечението с хелатиране на мед може да отнеме до шест месеца, за да започне да действа, и тези терапии могат да помогнат за справяне с атаксия, дистония и тремор, както и да предотвратят развитието на контрактури, които могат да бъдат резултат от дистония.
Трансплантация
Чернодробната трансплантация е ефективен начин за лечение на болестта на Уилсън, но се използва само в определени случаи поради рисковете и усложненията, свързани с процедурата. Използва се главно при хора с фулминантна чернодробна недостатъчност, които не реагират на медикаментозно лечение, или при хора с напреднало хронично чернодробно заболяване. Трансплантацията на черен дроб се избягва при тежки нервно-психични заболявания, при които ползата от нея не е доказана.
Усложнения
Основните усложнения при пациенти с нелекувана болест на Wilson са остра чернодробна недостатъчност, хронична чернодробна дисфункция с портална хипертония или хепатоцелуларен карцином, а понякога необратим прогрес към цироза, която се характеризира с прогресираща отпадналост, умора, анорексия, жълтеница, паякообразни ангиоми, спленомегалия (увеличен далак/слезка) и асцит.
Кървене от варици, чернодробна енцефалопатия, хепаторенален синдром и аномалии на коагулацията се появяват при чернодробна недостатъчност.
Смъртта настъпва обикновено около 30-годишна възраст, ако не се извърши спешна чернодробна трансплантация.
При болестта на Wilson се наблюдават и психични нарушения в 10%-20% от случаите, като клиничните прояви се обхващат диапазон от поведенчески нарушения през двигателни разстройства (понякога хореоатетозни) до паркинсонови прояви.
Прогноза
Ако не се лекува, болестта на Уилсън има тенденция да се влошава прогресивно и в крайна сметка да доведе до фатален изход.
Сериозните усложнения включват чернодробна цироза, остра бъбречна недостатъчност и психоза. Може да се развие карцином на черния дроб и холангиокарцином, но с по-ниска честота в сравнение с други хронични чернодробни заболявания.
С ранно откриване и лечение повечето от засегнатите могат да живеят относително нормален живот и да имат очаквана продължителност на живота, близка до тази на общото население. Чернодробните и неврологичните увреждания, възникнали преди лечението, могат да се подобрят, но често са постоянни. Фертилитетът обикновено не е засегнат и усложненията при бременност не се увеличават при пациенти с болест на Уилсън, която се лекува.
Симптоми и признаци при Разстройства на обмяната на медта МКБ E83.0
ВсичкиЛечение на Разстройства на обмяната на медта МКБ E83.0
Изследвания и тестове при Разстройства на обмяната на медта МКБ E83.0
Продукти от Framar.bg

КУПРУМ МЕТАЛИКУМ 9 СН

КУПРУМ МЕТАЛИКУМ 15 СН

Коментари към Разстройства на обмяната на медта МКБ E83.0