Spina bifida МКБ Q05
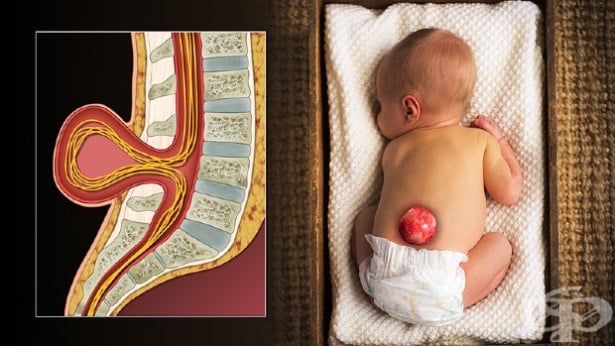
Под термина спина бифида — spina bifida (от латински: "разделен на две/разцепен гръбначен стълб") се разбира вроден дефект, при който дъгата на някои от прешлените на гръбначния стълб е или непълно затворена, или въобще липсва.
Мембраните около гръбначния мозък също не са напълно затворени.
Въпреки че този дефект е наричан още миелодисплазия (myelodysplasia) и миеломенингоцеле (myelomeningocele), повечето специалисти използват термина spina bifida.
Spina bifida е лечима гръбначна малформация, проявяваща се с различна степен на тежест.
Най-честата локализация на дефекта е долната част на гърба, но в редки случаи може да е по средата на гърба или в областта на врата.
Това състояние, класифицирано като дефект на невралната тръба (ембрионалната структура, от която се развиват главния и гръбначния мозък), е описано още преди около 4000 години.
Дефектите на невралната тръба се проявяват по различен начин — от раждане на мъртво дете до случайно откриване на spina bifida occulta (спина бифида окулта) при рентгеново изследване по друг повод.
Още при раждането може да се установи миеломенингоцеле — една от формите на спина бифида.

Изображение: https://tspace.library.utoronto.ca
При пациентите с миеломенингоцеле се установяват голям набор от увреждания, като основните функционални дефицити са:
- парализа на долните крайници;
- сетивни нарушения;
- дисфункция на пикочния мехур и червата;
- когнитивни нарушения;
За откриване на дефекти на невралната тръба се използват кръвни тестове, амниоцентеза или и двете.
Тези методи на изследване обикновено се използват в комбинация с фетална ехография (фетус — плод).
Епидемиология
Средната честота на спина бифида в световен мащаб е 1 случай на 1000 раждания.
Средната честота в развитите страни е около 0.4 случая на 1000 раждания.
Смята се, че част от тази разлика се дължи на факта, че бялата раса е с по-висок риск за развитие на това заболяване, както и на фактори на околната среда и географското разположение.
Честотата на този дефект е малко по-висок при жените, отколкото при мъже (1.2:1).
В много популации ниският социално-икономически статус се свързва с по-висок риск от дефекти на невралната тръба.
При около 5% от хората е установена спина бифида окулта (spina bifida occulta).
Най-високи стойности на този дефект са установени в някои части на Британските острови, основно Ирландия и Уелс, където през 1970 година са били докладвани 3-4 случая на миеломенингоцеле на 1000 души население, както и повече от 6 случая на аненцефалия на 1000 души население.
Етиология
 Смята се, че етиологията на spina bifida е многофакторна, включваща генетични и расови фактори, както и фактори на околната среда, сред които храненето има ключово значение — дефицитът на фолиева киселина (витамин В9) по време на бременност има огромно значение за развитие на този дефект.
Смята се, че етиологията на spina bifida е многофакторна, включваща генетични и расови фактори, както и фактори на околната среда, сред които храненето има ключово значение — дефицитът на фолиева киселина (витамин В9) по време на бременност има огромно значение за развитие на този дефект.
Цитоплазмени фактори, полигенетично унаследяване, хромозомни аберации и тератогенни фактори също са били разглеждани като възможни причини.
Малък брой от случаите са свързани с конкретни етиологични фактори.
Ако се роди дете с този дефект или ако някой от родителите го има, съществува 4% риск следващото дете също да бъде със спина бифида.
Майките на повечето бебета с миеломенингоцеле нямат други деца с малформации.
Въпреки това, другите поколения в семейство с 1 засегнато дете са с по-висок риск за развитие на дефекти на невралната тръба, отколкото са децата без засегнати братя и сестри.
Рискът е 1 на 20-30 последващи бременности, а ако са засегнати деца — рискът става 1 на 2 бременности.
Увеличен риск за развитие на миеломенингоцеле се съобщава и за роднини от втора и трета степен на засегнатите лица.
При около 10% от фетусите с установен в началото на бременността дефект на невралната тръба се установява хромозомна аномалия, включително тризомия 13 и 18, триплоидия и мутации в единични гени (моногенни мутации).
При жени с прегестационен диабет (развил се преди бременността) рискът за раждане на дете с малформация на централната нервна система (ЦНС), включително миеломенингоцеле, е 2-10 пъти по-висок в сравнение с общата популация.
Рискът при жени, които развиват гестационен диабет, е по-нисък в сравнение с риска при жени с прегестационен диабет.
Други рискови фактори за миеломенингоцеле включват:
- майчино затлъстяване;
- хипертермия (повишена телесна температура) в резултат на треска или фебрилно състояние, или при използване на сауни, джакузита или солариуми;
- диария;
- лошо контролиран диабет;
Други установени рискови фактори включват също така интраутеринно (вътрематочно) въздействие на антиепилептични медикаменти, особено валпроат и карбамазепин, както и лекарства, използвани за индуциране на овулация (изкуствено предизвикан растеж на фоликули).
Рискът за раждане на дете с дефект е повишен и при:
- майчина експозиция на фумонизини;
- електромагнитни полета;
- токсични отпадъци;
- продукти за дезинфекция;
- открити в питейната вода;
- пестициди;
Изображение: www.freepik.com
Патофизиология
Патофизиологично, дефектите на невралната тръба са резултат от тератогенен процес, в резултат на който се нарушава нормалното затваряне на ембрионалната неврална тръба, както и нейното нормално диференциране.
Дефектите на невралната тръба възникват между 17-ти и 30-ти ден на бременността, като много често жената дори не знае, че е бременна.
При нормални обстоятелства затварянето на невралната тръба настъпва около 23 и 27 ден след оплождането.
Ако са налице фактори, възпрепятстващи правилното затваряне на тръбата, настъпва дефект на невралната тръба.
Най-честите дефекти на невралната тръба са аненцефалия и миеломенингоцеле.
Някои форми на този дефекти възникват в резултат на първични състояния, които водят до повишено налягане в централната нервна система (ЦНС), което е предпоставка за комбинирана патогенеза.
 Изображение: www.freepik.com
Изображение: www.freepik.com
Някои медикаменти (антиконвулсанти), диабет, затлъстяване и наличието на роднина със spina bifida могат да увеличат риска за възникване на малформации на невралната тръба.
Затлъстяването е широко разпространено при деца с миеломенингоцеле, особено при лезии в лумбалната (поясната) и гръдната област, поради ограничен калориен разход.
Намалената мускулна маса в долните части на тялото води до намаляване на базалния метаболизъм.
Наднорменото тегло като цяло нарушава независимостта и придвижването на пациента.
При пациенти с миеломенингоцеле е намалена костната минерална плътност.
При децата с миеломенингоцеле е увеличен рискът за фрактури (счупвания) на долните крайници.
Намалената мускулна активност в засегнатия крайник и намалената възможност за натоварване води до намаляване на костната маса.
Много фрактури възникват след ортопедични интервенции.
При пациентите със спина бифида се установяват нарушения в отделителната система.
Основният фактор, определящ нарушенията в горния уринарен тракт, е промяната на интравезикалното (в пикочния мехур) налягане при задържането и отделянето на урината.
Момичетата с миеломенингоцеле навлизат в пубертета 1-2 години по-рано, отколкото техните незасегнати връстници.
Преждевременно полово развитие се свързва с хидроцефалия и затлъстяване.
Наблюдава се намалена генитална чувствителност, но някои жени с менингоцеле са в състояние да достигнат до оргазъм.
Фертилитетът (плодовитостта) при жени с миеломенингоцеле не е засегнат, но при бременните е налице:
- повишен риск от инфекция на пикочните пътища;
- болки в гърба;
- отслабване на тонуса на мускулите на тазовото дъно след раждането;
Младите мъже с миеломенингоцеле са с:
- нарушена генитална чувствителност;
- намалена способност за достигане и поддържане на ерекция;
- намален фертилитет;
Въпреки това обаче възможността за еякулация и възпроизвеждане трябва да се преценява за всеки отделен пациент.
Класификация
Класификацията на този дефект в развитието включва:
Spina bifida cystica (Спина бифида цистика)
 Спина бифида цистика може да възникне навсякъде по протежението на гръбначната ос, но най-често се открива в лумбалната (поясната) област.
Спина бифида цистика може да възникне навсякъде по протежението на гръбначната ос, но най-често се открива в лумбалната (поясната) област.
При тази форма на спина бифида гръбначният стълб е разцепен на две и се формира киста.
През дефекта на прешленните дъги излиза кистозно образувание на мозъчните обвивки — менингоцеле.
При хората с менингоцеле могат да не се наблюдават и неврологични усложнения.
Spina bifida cystica може да създаде проблеми, ако в менингоцелето навлезе мозъчна тъкан — в този случай кистата се нарича миеломенингоцеле, известна също и като отворена спина бифида.
Миеломенингоцеле е най-значимата и най-срещаната форма на spina bifida, като се среща в около 94% от случаите (тази цифра не включва spina bifida occulta).
Дете, родено с миеломенингоцеле, изисква специални грижи и преместване в специализирано звено, където може да се извърши неонатална хирургична намеса с последващо затваряне на дефекта.
Друга форма (най-тежката форма) на spina bifida cystica е миелоцеле или миелосхизис (myeloschisis) — разновидност, при която засегнатата област е във вид на сплескана, подобна на пластина маса от нервна тъкан, покрита вторично от епител.
Изображение: emedicine.medscape.com
Spina bifida occulta (спина бифида окулта)
 Името на тази форма идва от латинския термин "occulta", означаващ "скрит".
Името на тази форма идва от латинския термин "occulta", означаващ "скрит".
Това е най-леката форма на спина бифида и не включва пълните характеристики на spina bifida, като може да се наблюдава и при голям брой възрастни индивиди.
При спина бифида окулта дъгата на някои от прешлените не е напълно затворена, но дефектът е толкова малък, че гръбначният мозък не излиза навън.
Кожата на мястото на лезията може да бъде нормална, може да е налице леко окосмяване, може да има трапчинка или петно на това място.
Много от хората с този дефект дори не знаят, че го имат, тъй като не се установяват симптоми.
Честотата на spina bifida occulta е приблизително 10-20%, като повечето пациенти биват диагностицирани случайно при образно изследване на гръбначния стълб по друг повод.
Изображение: https://anndermatol.org
Сирингоменингоцеле (syringomeningocele)
Сирингоменингоцеле е друга форма на спина бифида.
С този термин се описва кухина, оградена от мембрана, в която има много малко количество гръбначен мозък.
Кухината се свързва централния канал на гръбначния мозък посредством гръбначномозъчната течност (ликвор).
Сирингомиелоцеле (syringomyelocele)
") Сирингомиелоцеле е форма на spina bifida, при която протрузията (издаването напред) на мембраните и гръбначния мозък води до значително увеличаване на количеството ликвор в централния канал, така че мозъчната тъкан образува тънкостенна торбичка (сак), която излиза през незатворените прешленни дъги.
Сирингомиелоцеле е форма на spina bifida, при която протрузията (издаването напред) на мембраните и гръбначния мозък води до значително увеличаване на количеството ликвор в централния канал, така че мозъчната тъкан образува тънкостенна торбичка (сак), която излиза през незатворените прешленни дъги.
Сирингомиелия (syringomyelia)
")
Сирингомиелия или хидросирингомиелия представлява наличието на кухини в гръбначния мозък, които може да са възникнали в резултат от разпадането на глиоматозни структури.
Изображение: www.mayoclinic.org
Клинична картина
Признаците и симптомите на спина бифида се установяват при раждане или пренатално (преди раждането).
При раждане, по срединната линия на гръбначния стълб се установява дефект в задната част на прешлените, с протрузия на менингите (мозъчните обвивки) и невралните елементи през дефекта.
Физически проблеми
Физическите проблеми на пациенти със spina bifida включват:
- слабост и парализа на долните крайници;
- ортопедични аномалии (дислокация на тазобедрената става, сколиоза, talipes equinovarus);
- нарушения в нормалното функциониране на пикочния мехур и червата, включително инконтиненция (незадържане), инфекции на пикочните пътища и нарушена бъбречна функция;
- декубитални рани и възпаление на кожата;
- патологични движения на очите;
При около 68% от децата със спина бифида се установява алергия към латекс, варираща от лека до животозастрашаваща.
Честата употреба на латекс в медицинските заведения увеличава значително рисковете от алергични състояния.
Най-срещаният подход да се предотврати развитието на алергия е да се ограничи контакта с латекс-съдържащи продукти, като например ръкавици и катетри.
Наличието на гръбначномозъчна лезия или белези вследствие на операция може да доведе до синдром на фиксирания (закотвен) гръбначен мозък (Tethered cord syndrome).
При някои пациенти това води до значителна тракция (придърпване) и натоварване на мозъчното вещество, което може да доведе до:
- влошаване на налична парализа;
- сколиоза;
- болки в гърба;
- влошаване на функцията на червата и/или пикочния мехур;
Неврологични проблеми
 При много индивиди със спина бифида се наблюдава и придружаваща аномалия на малкия мозък, наречена малформация на Арнолд-Киари тип II (Arnold-Chiari II).
При много индивиди със спина бифида се наблюдава и придружаваща аномалия на малкия мозък, наречена малформация на Арнолд-Киари тип II (Arnold-Chiari II).
При тези пациенти част от малкия мозък (малкомозъчните тонзили) е изместена от задната част на черепната кухина надолу през големия тилен отвор (foramen magnum), намиращ се в основата на черепа.
При около 90% от хората с миеломенингоцеле е налице и хидроцефалия, защото изместването на малкия мозък пречи на нормалната циркулация на гръбначномозъчната течност, което води до нейното натрупване.
При хора със спина бифида малкият мозък обикновено е с по-малки размери от нормалното, особено при пациенти с по-силно изразена лезия.
Мазолестото тяло (corpus callosum) е непълно развито в 70-90% от случаите с миеломенингоцеле — това се отразява на процесите на комуникация между лявото и дясното мозъчно полукълбо.
Могат да се наблюдават и аномалии на мозъчната кора, както и промяна в мястото на невроните в кората на главния мозък.
Изображение: https://radiopaedia.org
Нарушения в изпълнителните функции
Някои проучвания са установили трудности с изпълнителните функции на органите при млади индивиди със spina bifida.
За разлика от нормално развиващите се деца, при младите хора със спина бифида не се установява подобрение на изпълнителните функции с напредване на възрастта.
Конкретните области на затруднение при някои индивиди включват:
- планиране, организиране, иницииране на поведението;
- нарушения в паметта;
Способностите за решаване на проблеми, абстрактното мислене и визуалното планиране също могат да бъдат нарушени.
Освен това, при засегнатите деца може да се наблюдават нарушения в когнитивните функции.
Нарушения на вниманието
При индивидите със спина бифида могат да се наблюдават проблеми с обучението, особено по математика и четене.
Освен от аномалиите, засягащи мозъчната тъкан, които са пряко свързани с академичните умения, нарушенията в обучението се дължат и на нарушеното внимание и контрол на изпълнителните функции.
Децата със спина бифида могат да се представят добре в началното училище, но започват да изпитват затруднения в по-горните класове.
Диагноза
Диагностицирането на дефектите на невралната тръба включва различни лабораторни и инструментални методи:
Лабораторни скринингови тестове
Използват се за установяване на дефекти на невралната тръба и могат да се извършват чрез кръвни тестове, амниоцентеза или и двете.
Те обикновено се използват в комбинация с фетална ехография.
Наличието на отворен дефект на невралната тръба по време на ембрионалното развитие се характеризира с повишено ниво на алфа-фетопротеин (AFP) в амниотичната течност.
Пикови концентрации на AFP между 13 и 15-та седмица на бременността насочват към диагнозата, като ехографско потвърждение и извършване на амниоцентеза обикновено е възможно между 15 и 18 седмица.
Малко вероятно е чрез изследване на алфа-фетопротеин да се открие енцефалоцеле или покрито с кожа миелоцеле.
При деца със спина бифида, в допълнение към рутинните лабораторни скрининг тестове, се изследват и:
- нивата на антиконвулсанти в кръвта;
- урокултура;
- в някои случаи и определяне на кожната чувствителност към латекс;
Изследване на урина
Извършва се анализ на урината, урокултура и изследване на креатинин и урея в серума, за да се направи оценка на бъбречната функция при новородено със спина бифида.
Препоръчват се редовни урокултури при деца с везикоуретерален рефлукс или признаци и симптоми на инфекция на пикочните пътища.
Чрез микционна цистография се прави оценка на долните пикочни пътища, включително капацитетът на пикочния мехур и наличието на везикоуретерален рефлукс.
Изследване на алфа-фетопротеин и ацетилхолинестераза
Нивата на алфа-фетопротеин в кръвта на майката се изследват в началото на второто тримесечие.
Стойностите на AFP са повишени в 70-75% от случаите, в които се установява отворена spina bifida на плода.
Диагнозата се потвърждава чрез амниоцентеза и изследване на околоплодната течност за алфа-фетопротеин, както и за наличието на ацетилхолинестераза.
Миеломенингоцеле може да бъде открито в около 90% от засегнатите фетуси (фетус - плод) чрез комбинираното използване на тези тестове.
Ако дете има дефект на невралната тръба, то следващите деца са с повишен риск за развитие на този дефект.
Ето защо след раждане на дете със спина бифида се препоръчва амниоцентеза по време на последващите бременности, за да се проследи нивото на AFP.
Психометрично изследване
 Психометричните оценки на интелекта и познавателните функции са показани при пациенти с хидроцефалия, както и при пациенти, при които се установява дефицит в речта и езиковите функции и/или когнитивните или академичните умения.
Психометричните оценки на интелекта и познавателните функции са показани при пациенти с хидроцефалия, както и при пациенти, при които се установява дефицит в речта и езиковите функции и/или когнитивните или академичните умения.
Изображение: User:Life of Riley, CC BY-SA 3.0, via Wikimedia Commons
Анализ на походката
 Анализът на походката се използва, за да се оцени функционалното състояние на пациента.
Анализът на походката се използва, за да се оцени функционалното състояние на пациента.
Използва се и за изследване на инервацията на мускулите, силата и координацията им, тъй като нарушенията могат да нарушат способността за ходене или способността на пациентите да живеят самостоятелно.
Изображение: spinabifida.org
Фетална ехография
 Комбинацията от скринингово изследване на алфа-фетопротеин в майчиния серум и ултразвуково скринингово изследване през втория триместър на бременността спомага за откриване на над 90% от дефектите на невралната тръба след 20 гестационна седмица.
Комбинацията от скринингово изследване на алфа-фетопротеин в майчиния серум и ултразвуково скринингово изследване през втория триместър на бременността спомага за откриване на над 90% от дефектите на невралната тръба след 20 гестационна седмица.
Диагнозата "миеломенингоцеле" е сигурна, когато са налице следните 3 класически признака:
- вдлъбнатина на фронталните кости;
- вентрикуломегалия (патологична промяна, при която е налице увеличен размер на мозъчните стомахчета);
- малформация на Арнолд-Киари тип II;
Понастоящем ехографията не е достатъчно чувствителен метод, който да осигури надеждно и точно установяване на степента на развитие на дефекта.
Изображение: Wolfgang Moroder, CC BY-SA 3.0, via Wikimedia Commons
Компютърна томография (КТ) и ядрено-магнитен резонанс (ЯМР)
 Случаите с хидроцефалия могат да бъдат проследени чрез серийни краниални ехографии (при бебета) или чрез компютърна томография (КТ).
Случаите с хидроцефалия могат да бъдат проследени чрез серийни краниални ехографии (при бебета) или чрез компютърна томография (КТ).
КТ на глава е подходяща за оценка на възможността за рецидивираща хидроцефалия или промяна в размера или функцията на вентрикулите (мозъчните стомахчета).
Магнитен резонанс (ЯМР) на гръбначния стълб и мозъка е полезен за неврологична оценка.
ЯМР предоставя много детайлна информация за гръбначния мозък и неговите малформации.
Изображение: www.researchgate.net, Creative Commons Attribution-NonCommercial 4.0 International
Рентгенография
 Рентгенографиите на гръбначния стълб осигуряват информация за ранна оценка на състоянието, когато бебето е родено с миеломенингоцеле.
Рентгенографиите на гръбначния стълб осигуряват информация за ранна оценка на състоянието, когато бебето е родено с миеломенингоцеле.
С помощта на конвенционална рентгенография се определя степента на дефекта.
Вродените гръбначни изкривявания трябва да бъдат проследявани внимателно.
Обикновените рентгенографии са важни за клинична оценка на пациентите със сколиоза, дисплазия и дислокация на тазобедрената става.
Рентгенографията, заедно с ехографско изследване, трябва да се използва за оценка на всяка област на болка поради високия риск от патологични фрактури (счупвания).
Изображение: Wikipedia, Public domain, via Wikimedia Commons
Лечение
Хирургично лечение
 Стандартното лечение на spina bifida е хирургичното коригиране на дефекта след раждането, като целта на операцията е да се предотврати по-нататъшно увреждане на нервната тъкан и да се предотврати развитието на инфекция.
Стандартното лечение на spina bifida е хирургичното коригиране на дефекта след раждането, като целта на операцията е да се предотврати по-нататъшно увреждане на нервната тъкан и да се предотврати развитието на инфекция.
По време на операцията гръбначният мозък и неговите нервни коренчета се връщат на нормалното им анатомично място.
Като допълнение може да се постави шънт, с което да се осигури дрениране на излишната цереброспинална (гръбначномозъчна) течност, произведена в мозъка (както е при хидроцефалия).
Ако дефектът се установи по време на бременност, стандартното лечение се провежда след раждането.
Лечението на спина бифида преди раждането става по два начина:
- отворена фетална хирургия (хирургия на плода), при която матката се отваря и дефектът се коригира;
- посредством фетоскопия;
Лечението на spina bifida по време на бременност обаче крие рискове.
За майката те включват белези по матката, а за бебето има риск от преждевременно раждане.
При 15-35% от пациентите с малформация на Арнолд-Киари тип II се налага оперативна намеса.
Потенциалните хирургични кандидати са пациенти с:
- намалена подвижност или парализа на гласните връзки;
- изразен стридор;
- апнея;
- белодробна аспирация;
- сензомоторни нарушения;
Първоначалното лечение включва контрол на хидроцефалията. Ако това не подобри симптомите, налага се хирургична корекция на Chiari малформацията.
Повечето деца с миеломенингоцеле ще се нуждаят от периодични оценки от различни специалисти:
- физиотерапевти — спомагат чрез специфични терапии, помощни средства или медикаменти за по-добра функционална активност и максимално социално интегриране;
- ортопеди — наблюдават растежа и развитието на кости, мускули и стави;
- неврохирурзи — осъществяват оперативни интервенции след раждането и коригират различни усложнения;
- невролози — лекуват и правят оценка на проблемите на нервната система;
- уролози — за справяне с проблеми на отделителната система;
- офталмолози — за оценка и лечение на усложнения, засягащи очите;
- ортотисти — спомагат за изработката на различни видове помощни средства, включително протези, патерици, проходилки и колички за подпомагане на придвижването;
Поради потенциалните трудности при прехода дете-възрастен, препоръчва се юношите със спина бифида и техните семейства да започнат подготовка за този преход около 14-16-годишна възраст, въпреки че този период може да варира в зависимост от когнитивните и физическите способности на подрастващите.
Самият преход трябва да стане бавно и постепенно.
Медикаментозно лечение
Лекарствените средства, които най-често се използват при спина бифида, са такива за лечение на дисфункция на неврогенен пикочен мехур.
Групите медикаменти, които често се прилагат, са:
- Антиспазматични средства, действащи върху уринарния тракт (оксибутинин хлорид, oxybutynin chloride);
- Антихолинергични средства (хиосциамин сулфат, hyoscyamine sulfate);
- Трициклични антидепресанти (имипрамин хидрохлорид, imipramine hydrochloride);
- Алфа-блокери (алфа-адренергични антагонисти) — теразозин (Terazosin), доксазозин мезилат (Doxazosin mesylate), алфузозин (Alfuzosin);
Превенция
Няма една-единствена причина за развитие на спина бифида, нито установен начин за цялостната превенция (предотвратяване) на този дефект.
Според различни проучвания, липсата на фолиева киселина е фактор в патогенезата на дефектите на невралната тръба, включително spina bifida.
Доказано е, че приемът на храни, богати на фолиева киселина, спомага за намаляване честотата на дефектите на невралната тръба с около 70%, като също така намалява тежестта на вече възникналите дефекти.
Не е известно как и защо фолиевата киселина има този ефект.
Източници на фолиева киселина са пълнозърнести храни, зърнени закуски, зрял боб, листни плодове и зеленчуци.
Жените, които вече са имали бебе със спина бифида или друг тип дефект на невралната тръба, или приемат антиконвулсивни медикаменти, трябва да приемат по-високи дози фолиева киселина (4-5 милиграма на ден).
Прогноза
Смъртността при бебета с миеломенингоцеле се увеличава през първата година от живота.
Честотата на смъртните случаи при нелекувани новородени варира между 90 и 100% според различни проучвания, като повечето загиват през първата година от живота си.
Смъртните случаи през първите 2 години от живота на нелекуваните деца обикновено са резултат от хидроцефалия или интракраниална инфекция.
Бебе на възраст 2 месеца с нелекувано миеломенингоцеле има само 28% шанс да доживее 7-годишна възраст.
Заглавно изображение: https://www.scientificanimations.com, CC BY-SA 4.0, via Wikimedia Commons
Видове Spina bifida МКБ Q05
Симптоми и признаци при Spina bifida МКБ Q05
ВсичкиЛечение на Spina bifida МКБ Q05
Изследвания и тестове при Spina bifida МКБ Q05
Продукти свързани със ЗАБОЛЯВАНЕТО

ДОКТОР'С БЕСТ ФУЛИ АКТИВ ФОЛАТ капсули 400 мкг * 90
ПРЕГНАКЕЪР сироп 200 мл ВИТАБИОТИКС

КАЛИВИТА НЮ ЛАЙФ таблетки * 120

ДЖАРОУ ФОРМУЛАС МЕТИЛ ФОЛАТ капсули 400 мкг * 60
АРОФОЛИКАЦИД таблeтки 400 мкг * 90 АРО ЛАЙФ

ЕЛЕВИТ капсули * 30 BAYER

ПРЕГНАКЕЪР ПЛЮС ОМЕГА - 3 таблетки * 28 + капсули * 28 ВИТАБИОТИКС

ПРЕГНАКЕЪР НОВА МАМА таблетки * 56 ВИТАБИОТИКС

СОЛГАР ПРЕНАТАЛ таблетки * 120

НЕЙЧЪРС ЕЙД ФОЛИЕВА КИСЕЛИНА таблетки 400 мкг * 90

МИО - ИНОЗИТОЛ + ФОЛИЕВА КИСЕЛИНА 120 г VIRIDIAN

Коментари към Spina bifida МКБ Q05