Остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб МКБ Q77
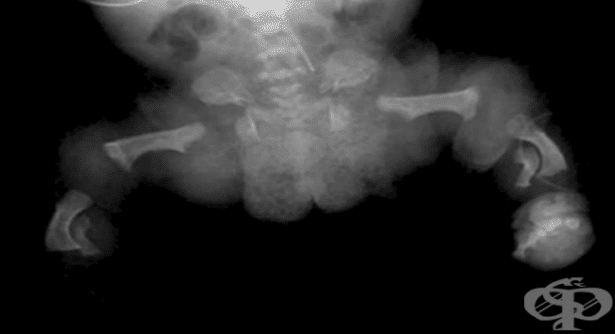
Тръбестите кости заедно с гръбначния стълб са едни от съставните части на скелета и мускулно-скелетната система. Скелетът представлява пасивната част на двигателния апарат и има няколко основни функции:
- защитна
- опорна
- хемопоетична
Остеохондродисплазия или скелетна дисплазия е общ термин за нарушение в развитието (дисплазия) на костите ("остео") и хрущяла ("хондро"). Това са редки заболявания с честота около 1 на 5 000 бебета, които водят до ограничение във функциите на организма, както и смърт.
Заболяванията от групата на остеохондродисплазиите главно се дължат на мутации в гени, кодиращи информацията за синтеза на белтъци, отговорни за клетъчния растеж, както и сформирането на костите и хрущялите.
Мутиралите гени се предават по автозомно-доминантен или рецесивен модел в поколенията, но е възможно мутациите да възникнат и съвсем случайно. Превенция и лечение за остеохондродисплазиите за съжаление няма.
 При заболяванията от групата на остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб, растежът и развитието на костите, хрущялите и съединителната тъкан са нарушени. Тези дефекти обикновено причиняват нисък ръст (джудже), аномалии в размерите на крайниците и други малформации. Някои видове дисплазии причиняват по-изразено скъсяване на крайниците, докато размерите на торса са незасегнати, а други причиняват основно съкращаване в дължината на торса, без да се наблюдават аномалии в крайниците.
При заболяванията от групата на остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб, растежът и развитието на костите, хрущялите и съединителната тъкан са нарушени. Тези дефекти обикновено причиняват нисък ръст (джудже), аномалии в размерите на крайниците и други малформации. Някои видове дисплазии причиняват по-изразено скъсяване на крайниците, докато размерите на торса са незасегнати, а други причиняват основно съкращаване в дължината на торса, без да се наблюдават аномалии в крайниците.
Остеохондродисплазиите са много на брой и се припокриват в някои клинични аспекти. Затова, рентгенологичното изследване е абсолютно необходимо, за да се установи точната диагноза. Диагностиката чрез ядрено-магнитен резонанс (ЯМР) може да осигури допълнителна информация и да насочи специалистите към правилната диагноза. Ранната диагностика и своевременната терапия на скелетната дисплазия са важни за борбата с функционалното влошаване.
Освен видимия нисък ръст, уголемения череп, изпъкналото чело и намаления обем на гръдния кош, се наблюдават и малформации в други органи и системи. Наблюдават се хидроцефалия, хипотония, дихателна недостатъчност, сърдечно-съдови проблеми, както и деформации на гръбначния стълб (сколиоза, кифоза и лордоза).
Средната височина на пациентите е около 130 см. В зависимост от вида дисплазия и степента на засягане на крайниците могат да се наблюдават различни изяви на аномалиите, причиняващи ниския ръст.
При някои пациенти се наблюдават скъсени крайници и нормален по размери торс, докато при други размерът на ръцете е нормален за сметка на намалените размери на торса, както и много други аномалии.
При децата с остеохондродисплазия се наблюдава забавено развитие на координационните способности и мускулно-скелетната система. Засегнатите прохождат и проговарят по-късно, в сравнение с деца, които не страдат от такова заболяване.
Слуховата обработка, краткотрайната и дълготрайната памет, както и вниманието са запазени, а умственото им развитие е нормално.
Значително подобряване на координацията и функциите при тези деца се наблюдава след петата година.
Диагностиката на остеохондродисплазиите се извършва главно чрез снемане на анамнеза, образни изследвания, както и кръвни изследвания.
Когато промените са налични още по време на ембрионалното развитие те могат да се диагностицират чрез извършване на антенатален (дородов) скрининг.
Всички изменения, които се манифестират по време на следродовия период се изследват чрез извършване на рентгенологични, ехографски, компютър-томографски и ядрено-магнитни изследвания.
Лечение за остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб няма. Терапията е насочена към облекчаване на някои от симптомите и подобряване качеството на живота на пациентите, както и към превенция на последваща неврологична и ортопедична симптоматика, която може да увреди състоянието на пациента.
При някои видове остеохондродисплазии се прилага и терапия със соматотропин. За съжаление хормонът има ефект при много малко видове остеохондродисплазии и не подобрява значително начина и качеството на живот на пациента.
Прогнозата за пациенти с остеохондродисплазия зависи от вида на заболяването, което я причинява. В зависимост от нарушенията и мутациите може да се наблюдават различни по вид аномалии, които в някои случаи да бъдат летални, а в други да не влияят на продължителността на живота.
Групата на остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб е обширна, а в нея се включват:
- Ахондрогенеза
- Нисък ръст (джудже)
- Синдром на късото ребро
- Chondrodysplasia punctata
- Ахондроплазия
- Дистрофична дисплазия
- Хондроектодермална дисплазия
- Спондилоепифизарна дисплазия
- Друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб
- Остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб, неуточнена
- Ахондрогенеза
 Ахондрогенезата представлява група от редки скелетни дисплазии, характеризиращи се със скъсяване в дължината на крайниците, малформации на прешлените, ребрата, гръдния кош и други части от скелета.
Ахондрогенезата представлява група от редки скелетни дисплазии, характеризиращи се със скъсяване в дължината на крайниците, малформации на прешлените, ребрата, гръдния кош и други части от скелета.
Ахондрогенезата се причинява от мутация в някой от следните гени: TRIP11; SLC26A2; COL2A1. В зависимост от това кой ген е мутирал, могат да се обособят три вида ахондрогенеза:
- тип 1 А (Хюстън-Харис тип)
- тип 1 В (Паренти-Фраккаро тип)
- тип 2 (Лангер-Салдино тип)
Ахондрогенезата се характеризира също с преждевременно раждане, прекомерно натрупване на течност в организма (hydrops fetalis) и изменения в главата - тя може да изглежда непропорционално голяма, поради малкия размер на тялото. В допълнение, засегнатите индивиди имат:
- скъсени крайници и ребра
- скъсен врат
- плоски прешлени
- неправилно развити кости на скелета
Здравословните проблеми, свързани с тези състояния, са животозастрашаващи и най-засегнатите бебета често са мъртвородени или умират скоро след раждането, поради тежката дихателна недостатъчност.
За диагностика на ахондрогенезата се използват образно-диагностични методи като рентгенография и ехография. Възможно е също да се вземе тъканна проба, за да се изследва под микроскоп (хистологично изследване) или да се извърши амниоцентеза.
Лечение за ахондрогенезата няма. Терапията е насочена срещу симптомите.
Повече за ахондрогенезата може да научите тук: Ахондрогенеза
- Нисък ръст (джудже)

Танатофорната дисплазия представлява тежко скелетно разстройство, характеризиращо се с изключително скъсени крайници и излишна кожа на ръцете и краката. Тя се включва като подвид на синдром на нисък ръст (джудже).
Тази аномалия е рядко наблюдавана. Среща се при 1 на 20 000-50 000 новородени.
Причината за това заболяване е мутация в гена на FGFR3 (Fibroblast Growth Factor Receptor 3). Този ген осигурява инструкции за създаване на протеин, който участва в изграждането и поддържането на костната и мозъчната тъкан.
Новородените с танатофорна дисплазия са най-често мъртвородени или умират скоро след раждането. Други особености на това състояние включват:
- тесен гръден кош
- къси ребра
- недоразвити бели дробове
- уголемена глава с голямо чело и широко разположени очи
Смъртността обикновено се дължи на тежка респираторна (дихателна) недостатъчност, поради намален гръден капацитет и недоразвити бели дробове и/или дихателна недостатъчност, дължаща се на компресия (притискане) на мозъчния ствол.
Диагностиката на това заболяване се извършва чрез внимателно снемане на анамнеза, както и някои лабораторни тестове. Лечение за танатофорната дисплазия няма.
Повече за танатофорната дисплазия и нисък ръст (джудже) може да научите тук: Нисък ръст (джудже)
- Синдром на късото ребро
 Синдромът на късото ребро представлява наследствено заболяване, което се характеризира с нарушения в ръста, полидактилия и други промени в тялото.
Синдромът на късото ребро представлява наследствено заболяване, което се характеризира с нарушения в ръста, полидактилия и други промени в тялото.
Този синдром е много рядко срещан, като се наблюдава при 1 на 100 000-130 000 новородени. Дължи се на мутация в гени IFT80 и DYNC2H1.
Най-честите изяви на синдрома на късото ребро са стесненият гръден кош, недоразвитите крайници и респираторната или бъбречната дисфункция, аномалиите в ребрата, както и чернодробните заболявания. Наблюдава се също така и белодробна хипоплазия (недоразвитие).
Индивидите, които преживеят периода след раждането и се справят с белодробната недостатъчност, в по-късен период развиват бъбречна или панкреасна недостатъчност, които от своя страна също водят до смърт.
Диагностиката се извършва чрез рентгенологични, ехографски и генетични изследвания.
Лечение за синдрома на късото ребро няма. Терапията е насочена към поддържането на живота и нормалната дихателна функция, поради тежкото състояние на дихателната система на новороденото.
Повече за синдрома на късото ребро може да научите тук: Синдром на късото ребро
- Chondrodysplasia punctata

Хондродисплазия пунктата представлява група от четири редки скелетни заболявания, които се проявяват с отлагане на калциеви соли в краищата на костите и хрущялите. Характерни за това заболяване са също и скъсяването на костите, отслабения мускулен тонус, нарушенията в зрението и слуха, както и плоското лице.
Chondrodysplasia punctata има четири форми, като честота на изява при всички тях е еднаква:
- Ризомелична хондродисплазия пунктата
- Х-свързана рецесивна chondrodysplasia punctata
- Х-свързана доминантна chondrodysplasia punctata
- Хондродистрофия калцификанс конгенита
Групата на chondrodysplasia punctata е с честота около 1 на 100 000 новородени. Заболяванията се унаследяват по автозомно-рецесивен начин, а прогнозата често е неблагоприятна.
Причиняват се от генни мутации, като в зависимост от мутиралия ген се проявяват различни признаци и изменения.
Диагностиката при заболявания от групата на хондродисплазия пунктата се извършва чрез образно-диагностични и лабораторни тестове, както и физикален преглед, заедно с внимателно снемане на анамнеза. В някои случаи рентгенографията може да бъде много полезна, защото добре визуализира калциевите отлагания в хрущялите и костите.
Лечението при chondrodysplasia punctata е насочено към индивидуалните симптоми и придружаващите заболявания. Възможно е да се наложи корекция на зрението или вродени сърдечни дефекти, както и физикална терапия, за да се подобри качеството на живот за пациентите с хондродисплазия пунктата.
Повече за chondrodysplasia punctata може да научите тук: Chondrodysplasia punctata
- Ахондроплазия
 Ахондроплазията представлява скелетно заболяване, характеризиращо се с нисък ръст (джудже), скъсени крайници, изменен гръден кош и промени в нервната, дихателната и сърдечно-съдовата система. При засегнатите индивиди, ръцете и краката са скъсени, докато торсът обикновено е с нормална големина.
Ахондроплазията представлява скелетно заболяване, характеризиращо се с нисък ръст (джудже), скъсени крайници, изменен гръден кош и промени в нервната, дихателната и сърдечно-съдовата система. При засегнатите индивиди, ръцете и краката са скъсени, докато торсът обикновено е с нормална големина.
Засегнатите обикновено имат средна височина от 131 сантиметра за мъжете и 123 сантиметра за жените. Другите характерни особености включват уголемена глава и чело.
В световен мащаб ахондроплазията е най-често срещаната скелетна дисплазия, засягаща около 1 на всеки 40 000 деца. Около 80% от всички "малки хора" имат ахондроплазия.
Приблизително 150 000 души страдат от аномалията в световен мащаб.
Ахондроплазията най-често се дължи на мутация в гена на фибробластния растежен фактор 3 (FGFR3). В около 80% от случаите мутацията се появява по време на ранното ембрионално развитие. В останалите 20% от случаите тя се наследява от родителите на индивида по автозомно-доминантен механизъм.
Диагностиката се извършва чрез образно-диагностични и физикални изследвания. Рентгенографията, ехографията и компютърната-томография имат голямо диагностично значение, защото чрез тях може да се потвърди диагнозата на лекуващия лекар при ранни симптоми на заболяването.
Лечението при ахондроплазия може да бъде както консервативно, така и хирургично. За консервативно лечение на това заболяване се използва главно соматотропин (човешки хормон на растежа).
Може да са необходими усилия за лечение или предотвратяване на усложнения като затлъстяване, хидроцефалия, обструктивна сънна апнея или инфекции на средното ухо.
Продължителността на живота на засегнатите лица е около 10 години по-малка от средната и могат да водят напълно нормален живот, ако имат необходимите условия.
Повече за ахондроплазията може да научите тук: Ахондроплазия
- Дистрофична дисплазия

Дистрофичната дисплазия се характеризира с нисък ръст и необичайно къси ръце и крака, анормално развитие на костите (скелетна дисплазия), както и на ставите в много области на тялото. Наблюдава се също така прогресивно абнормно изкривяване на гръбначния стълб (сколиоза или кифоза), необичайни промени в тъканта на външните видими части на ушите и малформации в областта на главата и лицето.
Заболяването засяга около 1 на 500 000 новородени в Съединените щати, което го прави много рядко.
Това състояние е по-често срещано във Финландия, където засяга около 1 на 33 000 новородени.
Дистрофичната дисплазия е едно от няколкото скелетни нарушения, причинени от мутации в SLC26A2 гена. Този ген осигурява инструкции за създаване на протеин, който е от съществено значение за нормалното развитие на хрущяла и за неговото превръщане в кост.
Индивидите страдащи от дистрофична дисплазия страдат от брахидактилия (необичайно къси пръсти), а ставите им е възможно да се сливат, което води до тяхното ограничено движение. Засегнатите бебета също така имат тежки деформации на стъпалата, дължащи се на аномалии и сливане на определени кости в тялото на всеки крак.
При повечето пациенти с дистрофична дисплазия има непълно затваряне на прешлените на гръбначния стълб (spina bifida occulta) и прогресивно необичайно странично изкривяване на гръбначния стълб (сколиоза).
Диагностиката на заболяването се осъществява чрез образно-диагностични и физикални изследвания. Възможно е да се извършат и кръвни или генетични изследвания, за да се диагностицира дисфункция на някои от органите или конкретния мутирал ген.
Лечението на дистрофичната дисплазия е насочено главно към симптомите и придружаващите заболявания. То е индивидуално и в зависимост от специфичните промени и изменения на пациента. Част от промените, освен консервативно се лекуват и хирургически, за да се постигне възможно най-добър ефект.
Повече за дистрофичната дисплазия може да научите тук: Дистрофична дисплазия
- Хондроектодермална дисплазия
 Хондроектодермалната дисплазия e рядко наследствено заболяване, водещо до нисък ръст. Характерни за аномалията са много скъсени кости на предмишницата, подбедрицата и ребрата. Заради аномалиите ръцете и краката изглеждат необичайно скъсени, защото размерите на торса и главата остават непроменени. Наблюдава се полидактилия (допълнителни пръсти) при засегнатите индивиди, както и други промени по външния им вид. Други характерни изменения при това заболяване са сърдечните и дихателните малформации, разцепване на устната и небцето, крипторхизъм, както и аномалии на зъбите.
Хондроектодермалната дисплазия e рядко наследствено заболяване, водещо до нисък ръст. Характерни за аномалията са много скъсени кости на предмишницата, подбедрицата и ребрата. Заради аномалиите ръцете и краката изглеждат необичайно скъсени, защото размерите на торса и главата остават непроменени. Наблюдава се полидактилия (допълнителни пръсти) при засегнатите индивиди, както и други промени по външния им вид. Други характерни изменения при това заболяване са сърдечните и дихателните малформации, разцепване на устната и небцето, крипторхизъм, както и аномалии на зъбите.
Честотата над хондроектодермалната дисплазия е 1:200 000.
Причината за това заболяване е мутация в гените EVC и EVC2. Тези генни мутации водят до производството на видоизменени EVC и EVC2 протеини. Смята се, че тези два протеина са важни за растежа и развитието. Някои засегнати индивиди нямат мутации в тези гени, така че е вероятно и други гени да носят отговорност за EVC протеините.
Хондроектодермалната дисплазия се наследява по автозомно-рецесивен модел.
Диагностиката на хондроектодермалната дисплазия се извършва чрез рентгенографско, компютър-томографско и ядрено-магнитно изследване. Генетични изследвания също може да бъдат проведени, за да се установи мутация в някой от двата гена (EVC, EVC2).
Лечението на заболяването зависи от структурните промени на тялото. То е насочено предимно към придружаващите симптоми и най-вече евентуалната дихателната недостатъчност, дължаща се на патологично намаленият обем на гръдния кош.
Повече за хондроектодермалната дисплазия може да научите тук: Хондроектодермалната дисплазия
- Спондилоепифизарна дисплазия

Спондилоепифизарната дисплазия е наследствено заболяване, което се характеризира с малформации на прешлените, дългите кости и ставите. Развива се поради дефект в структурните протеини на костите и хрущялните тъкани.
Заболяването има две форми в зависимост от мутиралите гени. Едната форма е автозомно-доминантна и се дължи на мутация в гена COL2A1, а другата е Х-свързана рецесивна форма, като се дължи на мутация в гена SEDL.
Двете форми на спондилоепифизарната дисплазия се различават по своите симптоми и начало на първите прояви. Най-честите симптоми са накуцване, болка в крайниците, умора и изкривявания на гръбначния стълб.
Диагностиката на заболяването се извършва чрез рентгенологично, ехографско или ядрено-магнитно образно изследване.
Лечението на тази дисплазия включва прилагането на ортопедични шини или извършването на операция за корекция на деформациите. Прилага се и терапия срещу другите симптоми на заболяването, както и терапия за укрепване на костната система.
Повече за спондилоепифизарната дисплазия може да научите тук: Спондилоепифизарната дисплазия
- Друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб
 Към групата на друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб се включват метатропната дисплазия, дисхондростеозата на Лери-Вайл, както и други заболявания като акромезомелична дисплазия, хондродисплазия с охлювоподобен таз и мезомелична дисплазия на Лангер.
Към групата на друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб се включват метатропната дисплазия, дисхондростеозата на Лери-Вайл, както и други заболявания като акромезомелична дисплазия, хондродисплазия с охлювоподобен таз и мезомелична дисплазия на Лангер.
Представляват нарушения в скелетната и хрущялна структура на тялото, като се проявяват с много нисък ръст, къси крайници, патологични изкривявания на гръбначния стълб, нарушения във функцията на органите и други. Всяко от заболяванията се проявява с различни аномалии, поради разликите в мутиралите гени.
Тези заболявания се дължат на мутации в гени, отговарящи за растежа и развитието на костната и хрущялна тъкан, и се срещат много рядко.
Диагностиката при тези заболявания се основава предимно на образни и клинични изследвания. На образните изследвания се наблюдават промени в костите и структурата на скелета. Извършват се често и молекулярно-генетични изследвания, които успешно идентифицират в кой ген е мутацията.
Лечението на заболяванията от тази група е насочено предимно към симптомите с цел подобряване начина на живот на пациентите. Възможно е да се приложи и хирургично лечение, за да се коригират по-тежки малформации.
Повече за друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб може да научите тук: Друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб
Видове Остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб МКБ Q77
Симптоми и признаци при Остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб МКБ Q77
- Затруднено дишане
- Нисък ръст
- Симптоми на челюстта
- Цепка на небцето
- Намален мускулен тонус
- Симптоми от пръстите
Библиография
https://emedicine.medscape.com/article/941176-overview
https://rarediseases.info.nih.gov/diseases/85/thanatophoricdysplasia
https://emedicine.medscape.com/article/945537-overview
https://rarediseases.org/rare-diseases/conradi-hunermann-syndrome/?fbclid=IwAR1d03JYKRq2IMcrkauPFSJLCnZjl6JL6pZ5fAYeWuM2N-eodQ4ePhIxI-E
https://rarediseases.org/rare-diseases/achondroplasia/
https://emedicine.medscape.com/article/1258401-treatment#showall
https://emedicine.medscape.com/article/1257787-overview
https://rarediseases.org/rare-diseases/ellis-van-creveld-syndrome/
https://rarediseases.info.nih.gov/diseases/4985/spondyloepiphyseal-dysplasia-tarda-x-linked
https://ghr.nlm.nih.gov/condition/spondyloepiphyseal-dysplasia-congenita
https://rarediseases.org/rare-diseases/metatropic-dysplasia-i/
Коментари към Остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб МКБ Q77