Chondrodysplasia punctata МКБ Q77.3
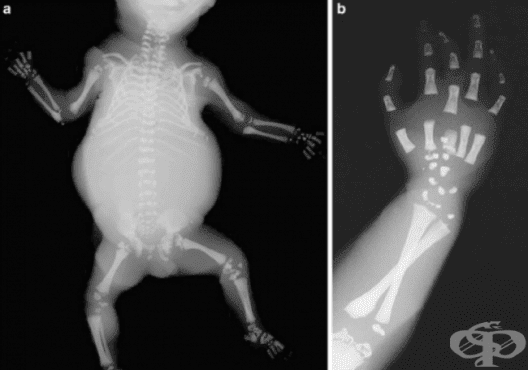
Под наименованието chondrodysplasia punctata се разбира група от скелетни заболявания, характеризиращи се най-често с отлагане на калциеви соли в хрущялите и краищата на костите. Тази група заболявания е с честота 1 на 100 000 живи раждания и се унаследяват по автозомно-рецисивен модел.
Chondrodysplasia punctata се характеризира със силно скъсяване на костите, забавяне в интелектуалното развитие, отслабен или усилен мускулен тонус, плоско лице, нарушение в зрението, суха кожа, която се лющи. Заболяването прогресира бавно.
Прогнозата при вродената аномалия е неблагоприятна.
Chondrodysplasia punctata има четири форми, като честота на всички тях е еднаква:
- Ризомелична хондродисплазия пунктата
- Х-свързана рецесивна chondrodysplasia punctata
- Х-свързана доминантна chondrodysplasia punctata
- Хондродистрофия калцификанс конгенита
- Ризомелична (rhizomelic) хондродисплазия пунктата (RCDP):
 Ризомеличната хондродисплазия пунктата е състояние, при което се наблюдава увреждане на нормалното развитие на много части на тялото. Основните признаци на това разстройство включват скъсяване на костите (ризомелия) на ръцете и краката, калцификации на междупрешленните дискове, наличие на малформации в телата на прешлените, увреждания в умственото развитие, замъгляване на очните лещи (катаракта) и респираторни проблеми. Това състояние се дължи на мутация в един от гените - PEX7, AGPS, GNPAT, FAR1, PEX5.
Ризомеличната хондродисплазия пунктата е състояние, при което се наблюдава увреждане на нормалното развитие на много части на тялото. Основните признаци на това разстройство включват скъсяване на костите (ризомелия) на ръцете и краката, калцификации на междупрешленните дискове, наличие на малформации в телата на прешлените, увреждания в умственото развитие, замъгляване на очните лещи (катаракта) и респираторни проблеми. Това състояние се дължи на мутация в един от гените - PEX7, AGPS, GNPAT, FAR1, PEX5.
Ставите при тези болни са болезнени, сковани и с ограничени движения. Теглото, дължината и обиколката на главата често са в долната граница на нормалното. Наблюдават се още припадъци, повторни инфекции на дихателни пътища, нарушения в зрението. Налице е умствено изоставане и повечето деца развиват гърчове.
Симптомите обикновено присъстват още при раждането или се появяват през първите няколко месеца от живота.
Описани са пет вида ризомелична хондродисплазия пунктата, класифицирани според генните мутации:
- тип 1 (RCDP1) с мутация на ген PEX7
- тип 2 (RCDP2) с мутация на ген GNPAT
- тип 3 (RCDP3) с мутация на ген AGPS
- тип 4 (RCDP4) с мутация на ген FAR1
- тип 5 (RCDP5) с мутация на ген PEX5
Всички тези гени участват във формирането и функционирането на клетъчни структури наречени пероксизоми. Пероксизомите са структури в клетката, които съдържат ензими, необходими за разграждането на различни вещества като мастни киселини и токсични вещества. В рамките на пероксизомите, протеините продуцирани от тези гени играят роля във формирането на липидни молекули наречени плазмалогени. Мутациите в тези гени предотвратяват формирането на плазмалогени в пероксизомите.
Дефицитът на мастни киселини и повишените концентрации на токсични вещества при засягане на пероксизомите уврежда растежа на костите.
 Диагнозата се поставя на базата клинични признаци и се потвърждава чрез биохимични генетични тестове (биохимичен скрининг) на пероксизомната функция, които включват — плазмена концентрация на фитинова киселина и мастни киселини, концентрация на червени кръвни клетки.
Диагнозата се поставя на базата клинични признаци и се потвърждава чрез биохимични генетични тестове (биохимичен скрининг) на пероксизомната функция, които включват — плазмена концентрация на фитинова киселина и мастни киселини, концентрация на червени кръвни клетки.
Лечение за ризомелична хондродисплазия пунктата няма. Терапията на пациентите включва физикална терапия, оперативно лечение на катарактата и ограничаване приема на фитинова киселина.
Прогнозата е неблагоприятна. Поради тежките си здравословни проблеми повечето хора с ризомелична хондродисплазия пунктата преживяват само няколко години. Рядко засегнатите могат да доживеят след 10–годишна възраст.
- Х-свързана рецесивна chondrodysplasia punctata
Х-свързаната рецесивна chondrodysplasia punctata е нарушение в развитието на хрущялите и костите, което се среща предимно в мъжкия пол.
При рентгенологично изследване се проявява като петна, разположени близо до краищата на костите и в хрущялите.
Други характерни особености на X-свързаната рецесивна хондродисплазия punctata включват нисък ръст, както и необичайно къси върхове и краища на пръстите.
Това състояние се свързва и с характерна форма на лицето и носа.
 Пациентите с X-свързана рецесивна хондродисплазия пунктата обикновено нямат проблеми с интелектуалното развитие и имат нормална продължителност на живота.
Пациентите с X-свързана рецесивна хондродисплазия пунктата обикновено нямат проблеми с интелектуалното развитие и имат нормална продължителност на живота.
От друга страна някои засегнати лица са имали сериозни или животозастрашаващи усложнения, включително необичайно задебеляване на хрущяла, образуващ дихателните пътища, което ограничава дишането и причинява стеноза на дихателните пътища.
Също така, аномалиите на шийните прешлени могат да доведат до притискане на гръбначния мозък, което да причини болка, скованост и слабост в мускулите на главата и шията.
Други, по-рядко срещани признаци на X-свързана рецесивна хондродисплазия пунктата включват забавено развитие, загуба на слуха, аномалии на зрението и сърдечни дефекти.
X-свързана рецесивна хондродисплазия пунктата е причинена от генетични промени, включващи ARSE гена. Този ген осигурява инструкции за получаване на ензим, наречен арилсулфатаза Е.
Функцията на този ензим е неизвестна, въпреки че изглежда важна за нормалното развитие на скелета и се смята, че участва в химичен път, включващ витамин К. Доказателствата показват, че обикновено витамин К има роля в растежа на костите и поддържане на костната плътност.
Между 60 и 75% от мъжете с характерните черти на X-свързана рецесивна хондродисплазия пунктата имат мутация в ARSE гена. Тези мутации намаляват или елиминират функцията на арилсулфатаза E.
Други 25% от засегнатите мъже имат малка делеция на генетичен материал от областта на X-хромозомата, която съдържа този ARSE ген. При тези мъже липсва целият ген, така че техните клетки не произвеждат функционална арилсулфатаза Е.
Все още не е доказано как недостигът на ензимът нарушава развитието на костите и хрущяла, характерно за X-свързана рецесивна хондродисплазия пунктата.
Както вече беше споменато заболяването се унаследява по автозомно-рецесивен начин. Генът, свързан с това състояние, се намира в X хромозомата, която е една от двете полови хромозоми.
При мъжете (които имат само една X-хромозома), едно изменено копие на ARSE гена във всяка клетка е достатъчно, за да причини състоянието.
При жените (които имат две X-хромозоми), трябва да се получи мутация и в двете копия на гена, за да се предизвика X-свързаната рецесивна хондродисплазия пунктата.
 Тъй като е малко вероятно жените да имат две изменени копия на този ген, мъжете са най-често засегнати от Х-свързани рецесивни разстройства. Характерно за рецесивното унаследяване е, че бащите не могат да предават Х-свързани аномалии на синовете си.
Тъй като е малко вероятно жените да имат две изменени копия на този ген, мъжете са най-често засегнати от Х-свързани рецесивни разстройства. Характерно за рецесивното унаследяване е, че бащите не могат да предават Х-свързани аномалии на синовете си.
Диагнозата се поставя чрез образно-диагностични и биохимични тестове - плазмена концентрация на арилсулфатаза Е, както и по наличието на кожни лезии.
Лечението X-свързаната рецесивна хондродисплазия пунктата е индивидуално и включва:
- физикална терапия
- корекция на зрението
- лечение на кожните проблеми
- предпазване от слънчевите лъчи
- корекция на вродените сърдечни дефекти и бъбречни аномалии
- честа оценка на кифосколиозата, която може да напредва бързо във всяка възраст
- Х-свързана доминантна chondrodysplasia punctata
Освен X-свързана рецесивна хондродисплазия пунктата съществува и друг вид X-свързана хондродисплазия пунктата, която от своя страна е доминантна. Другото име на това заболяване е синдром на Конради-Хюнерман (Conradi-Hünermann Syndrome).
Синдромът на Conradi-Hünermann е рядко генетично заболяване, характеризиращо се с:
- скелетни малформации
- аномалии на кожата
- катаракта
- нисък ръст
Специфичните симптоми и тежестта на заболяването могат да варират значително при отделните индивиди.
 Синдромът на Конради-Хюнерман се класифицира като форма на хондродисплазия пунктата, група от заболявания, характеризиращи се с образуването на малки калциеви петна, разположени по "нарастващата част" или главите на дългите кости (епифизи), както хрущялните структури в други области на тялото. Тези калциеви петна са видими след извършването на рентгенологични изследвания.
Синдромът на Конради-Хюнерман се класифицира като форма на хондродисплазия пунктата, група от заболявания, характеризиращи се с образуването на малки калциеви петна, разположени по "нарастващата част" или главите на дългите кости (епифизи), както хрущялните структури в други области на тялото. Тези калциеви петна са видими след извършването на рентгенологични изследвания.
Х-свързана доминантна хондродисплазия пунктата обикновено се свързва с непропорционално и асиметрично развитие на дългите кости, по-специално тези на горната част на ръцете и бедрената кост, малформации по хода на гръбначния стълб, както и слаб до умерен дефицит на растеж.
Синдромът на Conradi-Hünermann се унаследява по автозомно-доминантен модел и точно затова заболяването се среща почти изцяло при жените.
Симптомите, прогресията и тежестта на X-свързаната доминантна хондродисплазия пунктата могат да се различават драматично дори между членовете на едно и също семейство. Разстройството може да причини сериозни усложнения при раждането или от друга страна да бъде толкова леко, че симптомите да не се проявят до зряла възраст.
Важно е да се отбележи, че засегнатите лица не могат да имат всички изброени по-долу симптоми. Класическите симптоми на синдрома на Конради-Хюнерман включват увреждане на костната система, кожата и очите.
Засегнатите бебета може да не растат и да не наддават на тегло с очакваните темпове за възрастта и пола им. Забавеният растеж може в крайна сметка да доведе до нисък ръст през целия им живот.
В периода на ранно детство, индивидите с X-свързаната доминантна хондродисплазия пунктата имат малки, втвърдени калциеви отлагания по нарастващата част (епифизи) на дългите кости, както и други области от хрущялния скелет. Хрущялът е твърда и еластична съединителна тъкан, която осигурява опорна и структурна функция в тялото.
Когато скелетът започва да се развива, той се състои предимно от хрущял, който постепенно се заменя с кост. При кърмачета със синдром на Conradi-Hünermann се наблюдават калцификации в гръбначния стълб, таза, ребрата, гръдната кост, ключиците и в редки случаи ларинкса.
Хората с X-свързаната доминантна хондродисплазия пунктата обикновено имат допълнителни мускулно-скелетни аномалии. Такива белези често включват асиметрично съкращаване на дългите кости на крайниците, което води до асиметрия в дължината на ръцете и краката, като едната страна обикновено е по-засегната от другата. Възможно е също така да възникне скованост на ставите, както и малформации на бедрата (дисплазия на тазобедрената става), полидактилия, дефекти на гръбначния стълб и др.
Някои хора със синдром на Conradi-Hünermann могат да развият катаракта. Катарактата може да бъде вродена или да се развие по време на ранна детска възраст и да засяга едното или и двете очи. В редки случаи могат да се наблюдават и допълнителни очни аномалии, включващи:
- необичайно малки очи (микрофталмос)
- необичайно малки роговици (микрокорнея)
- бързо и неволево движение на окото (нистагъм)
В някои случаи очните аномалии значително намаляват зрението.
Допълнителни отличителни черти на лицето могат да включват:
- необичайно изпъкнало чело
- сплеснати скули
- плосък нос
- обърнати ноздри
- неправилна форма на ушите
В периода след раждане много от засегнатите бебета имат зачервяване (еритема) и необичайно удебеляване, сухота и лющене на кожата. В някои случаи засегнатите области на кожата могат да бъдат по-тъмни или по-светли от околните области (хипер- и хипопигментация). Части с развитие на косопад също могат да се наблюдават по скалпа (цикатрициална алопеция).
 В някои случаи се съобщава за допълнителни аномалии при синдрома на Конради-Хюнерман. Такива признаци включват стесняване на трахеята и/или ларинкса, необичайно къс врат, аномалии на ноктите, вродени сърдечни дефекти и други.
В някои случаи се съобщава за допълнителни аномалии при синдрома на Конради-Хюнерман. Такива признаци включват стесняване на трахеята и/или ларинкса, необичайно къс врат, аномалии на ноктите, вродени сърдечни дефекти и други.
Синдромът на Conradi-Hünermann се причинява от мутации в гена за емопамил-свързващия протеин. В много случаи тази мутация се появява случайно и се унаследява доминантно.
X-свързаните доминантни заболявания са състояния, причинени от анормален ген в Х-хромозомата. Жените имат две X-хромозоми, докато мъжете имат една X-хромозома и една Y-хромозома. При жените някои изменения на Х-хромозомата могат в някои случаи да бъдат "маскирани" от нормалния ген на другата X-хромозома. Въпреки това, тъй като мъжете имат само една X-хромозома, мутации свързани с Х-хромозомата, са по-вероятно да бъдат напълно изразени.
При мъже, които наследяват мутирал ген за X-свързано доминантно заболяване (хемизиготи), се подозира, че пълното изразяване на болестта може да бъде свързано със загуба на живота още преди раждането. В редки случаи са докладвани мъже със синдром на Конради-Хюнерман, придобит по време на ембрионалното развитие, а не унаследен от родителите или при мъже с анормален хромозомен брой.
Мъжете носители на това заболяване предават гена на дъщерите си, но не и на синовете си. Жените с копие на мутиралия ген имат 50% риск от предаване на гена на дъщерите и синовете си.
Емопамил генът създава протеин, известен като емопамил-свързващ протеин. Мутациите на емопамил гена водят до недостатъчни нива на функционални копия на този протеин. Емопамил-свързващият протеин функционира като ензим на стерол-8-изомеразата и играе роля в биосинтезата на холестерола. Клиничната картина на X-свързаната доминантна хондродисплазия пунктата се смята за свързана с натрупването на холестеролни прекурсори (стероли), които са токсични.
Диагнозата на синдрома на Conradi-Hünermann се прави чрез идентифициране на характерните симптоми, задълбочена клинична оценка и разнообразие от специализирани тестове.
Рентгенови, очни, кожни и биохимични изследвания могат да се извършат, за да се диагностицира X-свързаната доминантна хондродисплазия пунктата. Рентгенологичното изследване може да разкрие характерните изменения в епифизите и хрущялната тъкан.
Важен тест за потвърждаване на диагнозата на синдрома на Conradi-Hünermann е оценката на плазмената концентрация на стеролите. Стеролите са жизненоважни вещества за човешкия организъм. Те контролират пропускливостта на клетъчните мембрани и оказват влияние върху метаболитните процеси. Също така са съставна част са на липидите.
Мутациите на емопамил гена водят до натрупване на стероли в плазмата и някои тъкани на тялото. Нивата на стерола се измерват чрез газова хроматография (техника за разделяне на смеси, съставени от различни химически вещества).
Диагнозата на X-свързаната доминантна хондродисплазия пунктата може да бъде потвърдена и чрез молекулярно-генетично изследване, което може да идентифицира характерната генетична мутация, причиняваща нарушението.
Лечението на X-свързаната доминантна хондродисплазия пунктата е насочено към специфичните симптоми, които са видими при всеки индивид. Това лечение може да изисква съвместни усилия от множество медицински специалисти, които диагностицират и лекуват смущения на скелета, ставите, мускулите, кожата, очите и др.
 Хирургичните интервенции не са препоръчителни, освен в случаите на лицеви малформации, сколиоза и някои животозастрашаващи състояния. Представените хирургични процедури ще зависят от естеството, тежестта и комбинацията от анатомични аномалии, свързаните с тях симптоми и други фактори.
Хирургичните интервенции не са препоръчителни, освен в случаите на лицеви малформации, сколиоза и някои животозастрашаващи състояния. Представените хирургични процедури ще зависят от естеството, тежестта и комбинацията от анатомични аномалии, свързаните с тях симптоми и други фактори.
Препоръчителното лечение за вродена катаракта може да включва ранно хирургично отстраняване на измененията. За тези, които са засегнати от аномалии на кожата, могат да се препоръчат подходящи лекарства, които омекотяват и успокояват кожата.
Повечето хора с генетичното заболяване имат нормален живот. Някои от тях имат тежки медицински проблеми, които могат да доведат до ранна смърт — като например аномалии на дихателния тракт, които водят до стесняване на дихателните пътища или при аномалии на гръбначния стълб, които стесняват гръбначномозъчния канал, което притиска гръбначния мозък.
- Хондродистрофия калцификанс конгенита
Това заболяване е последното в групата на chondrodysplasia punctata и e известно под няколко имена. Името хондродистрофия калцификанс конгенита (Chondrodystrophia Calcificans Congenita) е дадено от Conradi, който описва първия си случай през 1914 г. и от Hünnermann през 1931 г.
 Болестта се проявява през първите дни или месеци от живота. Смъртността е висока през първата година, особено в по-тежките случаи. От друга страна пациентите само с няколко изразени симптома могат да водят напълно нормален живот и дори да покажат признаци на регресия (обратно развитие) на тези симптоми. Тези факти правят диагнозата доста трудна.
Болестта се проявява през първите дни или месеци от живота. Смъртността е висока през първата година, особено в по-тежките случаи. От друга страна пациентите само с няколко изразени симптома могат да водят напълно нормален живот и дори да покажат признаци на регресия (обратно развитие) на тези симптоми. Тези факти правят диагнозата доста трудна.
Честотата на заболяването е много ниска - 1 на 500000 раждания, а в описаните досега случаи се откриват повече болни жени, отколкото мъже.
Причините за заболяването не са напълно изяснени, но се предполага за автозомно-рецесивна мутация в някой от гените.
Основният симптом при хондродистрофия калцификанс конгенита е скъсяването на крайниците и дългите кости, като често засягането е несиметрично. Метафизите (междинния дял между диафизата и епифизата) на костите са широки, диафизите (средната част на дългите кости) са извити, наблюдава се кифоза, сколиоза, аномалии на пръстите и краката и др.
В около половината от случаите се наблюдават необичайни движения, ограничаване на движенията, болка при допир и др. Тези симптоми обикновено се отнасят за една или повече от по-големите стави - колянна, бедрена и лакътна. Дисфункцията на ставите се дължи не само на костните деформации, но и на промени в структурата на мускулите.
Тези симптоми обикновено се отнасят за една или повече от по-големите стави - колянна, бедрена и лакътна. Дисфункцията на ставите се дължи не само на костните деформации, но и на промени в структурата на мускулите.
Диагностицирането при това заболяване се извършва трудно и най-често се прави след провеждане на рентгенологично изследване. На рентгенологичния образ се наблюдават характерни калцификации по епифизите (заобленият край) на костите.
Тези калцификации са повече или по-малко равномерно разпределени по костите или ставната повърхност и могат да бъдат намерени във всяка кост на скелета.
Лечение за това заболяване няма. Терапията по-скоро е насочена към придружаващите заболявания или към симптомите на заболяването.
Симптоми и признаци при Chondrodysplasia punctata МКБ Q77.3
- Ментална ретардация
- Забавен растеж
- Нисък ръст
- Малка глава
- Симптоми на челюстта
- Намален мускулен тонус
Библиография
https://www.ncbi.nlm.nih.gov/books/NBK1270/
https://www.ncbi.nlm.nih.gov/books/NBK55062/
https://www.ncbi.nlm.nih.gov/books/NBK1544/
https://ghr.nlm.nih.gov/condition/rhizomelic-chondrodysplasia-punctata
https://ghr.nlm.nih.gov/condition/x-linked-chondrodysplasia-punctata-1
https://rarediseases.org/rare-diseases/conradi-hunermann-syndrome/?fbclid=IwAR1d03JYKRq2IMcrkauPFSJLCnZjl6JL6pZ5fAYeWuM2N-eodQ4ePhIxI-E
Коментари към Chondrodysplasia punctata МКБ Q77.3