Ахондроплазия МКБ Q77.4
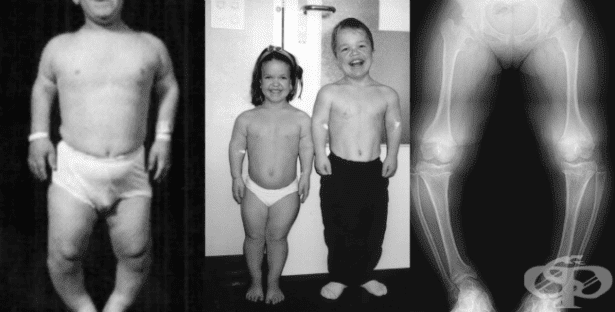
Скелетните дисплазии са група от заболявания, характеризиращи се с аномалии в растежа или развитието на хрущяла и костите. Те засягат черепа, гръбначния стълб и крайниците в различна степен.
Заболяванията, включени в тази група са:
- Ахондроплазия
- Леталната хондродисплазия на Блумстранд (Blomstrand lethal chondrodysplasia)
- Хондрогенезис имперфекта (хондрогенеза тип 2; Chondrogenesis imperfecta)
- Хипохондроплазия
- Нисък ръст (джудже)
- Остеосклерозис конгенита (вродена остеосклероза)
-
Ахондроплазия
Тя е най-честият вид заболяване от групата на скелетните дисплазии, изразяваща се с непропорционален нисък ръст и къси крайници (джуджета).
Терминът ахондроплазия, който означава липса или грешка във формирането на хрущял, е използван за пръв път през 1878 г. Въпреки че е неточен от хистопатологична гледна точка, неговата употреба е универсална и е приета в сферата на медицината.
 Аномалиите на ръста често се характеризират по това дали промените са по-изразени в крайниците или в гръдния кош.
Аномалиите на ръста често се характеризират по това дали промените са по-изразени в крайниците или в гръдния кош.
Ахондроплазията, хипохондроплазията и метафизните хондродисплазии се считат за аномалии, засягащи предимно крайниците, като костите на ръцете и бедрата се засягат по-често от костите на предмишниците, краката, дланите и ходилата.
Приблизително 10 000 души са засегнати от заболяването в Съединените щати. В световен мащаб ахондроплазията е най-често срещаната скелетна дисплазия, засягаща около 1 на всеки 40 000 деца. Около 80% от всички "малки хора" имат ахондроплазия.
Приблизително 150 000 души страдат от аномалията в световен мащаб.
Ахондроплазията се среща с еднаква честота при мъже и жени и се среща във всички раси с еднаква честота.
Причините за ахондроплазия могат да бъдат генетични и молекулярни.
От генетична гледна точка се наблюдава аномалия в четвърта хромозома. Заболяването се предава по автозомно-доминантен механизъм.
Около 80% от случаите са резултат от случайна нова мутация. Повечето родители са със среден ръст и нямат фамилна анамнеза за това заболяване. Рискът на родителите да имат второ засегнато от ахондроплазия дете е незначителен.
Когато и двамата родители страдат от ахондроплазия, има 50% вероятност следващото им дете да е засегнато, 25% да се случи спонтанен аборт през първите няколко месеца от бременността, а 25% шанс заболяването да не се предаде. Когато само един родител има ахондроплазия, шансът за предаване на този ген на всяко дете е 50%.
Освен генетични се наблюдават и молекулярни причини.
Факторите за растеж на фибробластите (Fibroblast growth factors-FGF) са протеини, свързани с клетъчния растеж, миграцията и зарастването на раните. На клетъчно ниво функцията на тези протеини се регулира от трансмембранни тирозин-киназни рецептори (FGFRs).
Мутация в някой от тези рецептори е отговорна за ахондроплазия и други видове дисплазии. Мутацията предизвиква  повишаване във функцията на рецепторите, което води до ограничаване на процеса на вкостяване.
повишаване във функцията на рецепторите, което води до ограничаване на процеса на вкостяване.
Основният дефект, открит при пациенти с ахондроплазия, е нарушение в процеса на вкостяване. Кухите кости са скъсени и разширени, костите на таза се изменят, а изграждането на някои хрущяли абнормално. По този начин ахондроплазията се изявява характерно клинично и рентгенологично.
Измененията при ахондроплазията са очевидни още при раждането. Диагнозата се извършва въз основа на физикален преглед и образно-диагностични изследвания.
Заболявания, свързани с ахондроплазия са:
- Рецидивиращ отит
- Неврологични усложнения, дължащи се на компресия (хипотония, респираторна недостатъчност, апнея, цианоза, квадрипареза, внезапна смърт)
- Обструктивни и ограничаващи респираторни усложнения
- Хидроцефалия
- Спинални (на гръбначния стълб) деформации (кифоза, лордоза, сколиоза)
- Стеноза (стеснение) на гръбначния канал
- Сърдечносъдови усложнения
При децата с ахондроплазия се наблюдава забавено развитие на координацията на тялото и в мускулно-скелетната им система. Засегнатите прохождат и проговарят по-късно, в сравнение с деца, които не страдат от това заболяване.
Когнитивните умения (слухова обработка, краткотрайна и дълготрайна памет, внимание) са запазени, както и умственото им развитие е в рамките на нормалното.
Значително подобряване на координацията и функциите при тези деца се наблюдава след петата година.
Средната височина на зрели пациенти е 130 см. Около 75% от тях страдат от чести възпаления на средното ухо, когато са на възраст под 5 години. Повтарящото се възпаление на средното ухо е често срещано, поради лошото оттичане на евстахиевите тръби, уголемяване на сливиците и временните костни аномалии. Тези чести възпалителни процеси са предпоставка за намаляване или пълна загуба на слуха.
 Обструкцията (запушване) на горните дихателни пътища, стесняването на гръдната стена и евентуалните неврологични проблеми от компресията (притискането) на мозъчния ствол намаляват жизнеността. Честотата на пневмониите, апнеята (липса на дишане) и други респираторни усложнения е увеличена. Симптомите на обструкция на дихателните пътища включват хъркане и спане в характерна позиция с изпънат врат.
Обструкцията (запушване) на горните дихателни пътища, стесняването на гръдната стена и евентуалните неврологични проблеми от компресията (притискането) на мозъчния ствол намаляват жизнеността. Честотата на пневмониите, апнеята (липса на дишане) и други респираторни усложнения е увеличена. Симптомите на обструкция на дихателните пътища включват хъркане и спане в характерна позиция с изпънат врат.
Неправилното развитие на основата на черепа води до стеснен тилен отвор (foramen magnum). Стесняването на форамен магнум компресира областта, в която мозъчния ствол преминава в гръбначния мозък, причинявайки симптоми на дихателна недостатъчност, апнея, проблеми с храненето, слабост в крайниците и внезапна смърт. Тези симптоми са чести през първите няколко години от живота. Хроничната компресия на мозъчния ствол също може да бъде причина за хипотония (понижено кръвно налягане), наблюдавана през първите 2 години от живота.
Стенозата на гръбначния канал и тилния отвор води до симптоми като болка в гърба, болка в краката, дизестезия (необичайно чувство за допир), парестезия (необичайни усещания, наподобяващи "мравучкане", парене, убождане и др.), долна парапареза (слабост в долните крайници) и др. Често болката се облекчава, ако пациентът клекне или се наведе напред.
Повече от 50% от пациентите изпитват симптоми на радикулопатия на долните крайници. Средната възраст на начало на симптомите е 26 години, а 1/3 от пациентите са на възраст около 15 години.
Ахондроплазията е очевидна още при раждането. Характеристиките включват:
- Разширен череп
- Изпъкнало чело
- Къса средна част на лицето
- Изявена долна челюст
- Гръдният кош е с намален обем, долните ребра са изгладени, а коремът е изпъкнал
Преди прохождането, детето има изразена кифоза в гръдния отдел на гръбначния стълб. Раменете изглеждат широки поради нормалното развитие на ключицата и добре развитата мускулатура. Късите ръце могат да допринесат за обемна мускулна маса и видимо повишена сила при такива индивиди.
Големият пищял (tibia) е изкривен, което води до значително деформиране на долните крайници. Походката обикновено е  клатушкаща.
клатушкаща.
Смъртността при това заболяване при деца под 4 години най-често се дължи на компресията на мозъчния ствол (причинява се внезапна смърт). При индивиди на възраст между 5 и 24 години аномалиите в централната нервна система (ЦНС) и дихателната система са най-често срещаните причини за смърт.
При лица на възраст 25-54 години сърдечносъдовите проблеми са най-честите причинители на леталитет.
За диагностика се използват предимно образно-диагностични и ДНК изследвания.
Ахондроплазията може да бъде открита още преди раждането чрез пренатална диагностика. ДНК тест също може да се извърши преди раждането, за да се открие евентуална хомозигозност, при която се наследяват две копия на мутиралия ген - летално състояние, водещо до мъртвородени бебета.
Изследването на скелета е полезно за потвърждаване на диагнозата. На рентгенологичния образ черепът е уголемен, с тесен тилен отвор и относително малка основа. Телата на прешлените са скъсени, сплескани и се наблюдава стеснен гръбначен канал. Кухите и дълги кости са скъсени и задебелени. Дланта е по-широка от нормалното с къси пръсти.
Ако не се наблюдават такива промени по скелета, диагнозата ахондроплазия трябва да се изключи и да се мисли в нова насока. Пренаталната диагностика може да бъде извършена чрез ултразвуково изследване, при което се наблюдава за несъответствие между дължината на бедрената кост и бипариеталния диаметър на черепа (диаметърът между двете теменни кости).
 Лечението при ахондроплазия може да бъде както консервативно, така и хирургично.
Лечението при ахондроплазия може да бъде както консервативно, така и хирургично.
За консервативно лечение на това заболяване се използва главно соматотропин (човешки хормон на растежа). Наличието му революционизира лечението на хора с нисък ръст. Понастоящем хормонът на растежа се използва за стимулиране на височината на пациентите с ахондроплазия. Най-голямото ускорение в скоростта на растежа се наблюдава през първата година от лечението и при тези с най-ниски скорости на растеж преди започване лечението.
Проучване от 2017 г. оценява увеличението в крайната височина при 22-ма възрастни пациенти с ахондроплазия. Установено е, че дългосрочното лечение със соматотропин е допринесло за увеличение от 2,6% в ръста на пациентите.
За максимална полза се препоръчва терапията да бъде започната на най-ранна възраст (1-6 години).
Повечето от ортопедичните проблеми, срещани при пациенти с ахондроплазия, са свързани с гръбначния стълб и налагат оперативно лечение.
Стенозата (стеснението) на големия тилен отвор и гръбначния канал, кифозата и деформациите на крайниците са част от ортопедичните проблеми, които често се отнасят до ахондроплазията.
Всички тези аномалии подлежат на хирургическа корекция, което да осигури по-добър начин на живот на пациента.
-
Летална хондродисплазия на Блумстранд (Blomstrand lethal chondrodysplasia)
Друго заболяване от групата на скелетните дисплазии е леталната хондродисплазия на Блумстранд.
Леталната хондродисплазия на Блумстранд е неонатална дисплазия, характеризираща се с костни малформации и аномалии, много къси крайници и пренатална смъртност.
Към днешна дата в литературата са описани по-малко от 10 случая.
Леталната хондродисплазия на Блумстранд е вродено заболяване, характеризиращо се с:
- ниско тегло при раждане
- лицеви аномалии (раздалечени и изпъкнали очи)
- катаракта
- малформации на носа
- постоянно изплезен език
- стеснен гръден кош
- сериозни малформации на крайниците
Други наблюдавани аномалии включват дефекти в развитието на зъбите, млечната жлеза, белите дробове и др.
Описани са две форми на това заболяване:
- Тип I - тежка, класическа форма
- Тип II - по-лека форма
Леталната хондродисплазия на Блумстранд се причинява от генетични мутации в гените, отговарящи за продукцията и действието на паращитовидните хормони.
 Признаците и симптомите на това заболяване могат да включват:
Признаците и симптомите на това заболяване могат да включват:
- Прекомерна калцификация на костите
- Генерализирана остеосклероза (увеличаване на костната плътност)
- Калцификация на ларинкса
- Микромелия (необичайно къси крайници)
Диагнозата се основава на клиничните изяви и образно-диагностичните методи, които показват генерализирано повишаване на костната плътност, тежко скъсяване на дългите кости с широки метафизи и анормални краища, издължен и стеснен гръден кош и др.
Хистопатологичното изследване показва засилено вкостяване на дългите кости, както и нарушения в сухожилията и части от костната тъкан. Диагнозата се потвърждава от биохимичен генетичен скрининг.
За съжаление няма лечение за този вид хондродисплазия, тъй като това е генетично заболяване. Терапията при леталната хондродисплазия на Блумстранд е насочена срещу признаците и симптомите, с цел подобряване начина на живот на пациента.
-
Хондрогенезис имперфекта (Chondrogenesis imperfecta)
Следващото заболяване от групата на ахондроплазиите е хондрогенезис имперфекта, още наричана ахондрогенеза тип 2 или синдром на Лангер-Салдино.
Децата с това заболяване имат къси ръце и крака, стеснен гръден кош с анормални ребра и недоразвити бели дробове. Това състояние е свързано и с липсата на вкостяване в костите, изграждащи гръбначния стълб и таза.
Отличителните черти, характерни за това заболяване включват:
- Изпъкнало чело
- Малка брадичка
- Цепнатина на небцето
- Подут корем
- Hydrops fetalis (излишък от течност, натрупана в тялото преди раждането)
Хондрогенезис имперфекта се среща сравнително рядко. В световен мащаб заболяването се наблюдава в 1 на 40 000 до 60 000 новородени.
Заболяването е едно от няколкото скелетни нарушения, които са резултат от мутации в гена COL2A1. Този ген осигурява инструкции за създаване на протеин, образуващ колаген тип II. Този тип колаген се намира предимно в хрущяла и очната ябълка (стъкловидното тяло). Това е от съществено значение за нормалното развитие на костите и съединителната тъкан.
Мутациите в гена COL2A1 пречат на сформирането на молекули за изграждане на колаген тип II, което пречи на правилното развитие на костите и съединителната тъкан.
Хондрогенезис имперфекта e автозомно-доминантно заболяване, тъй като само едно копие на мутиралия ген във всяка клетка е достатъчно, за да причини състоянието.
Измененията в генетичната информация почти винаги са причинено от нови мутации в гена COL2A1 и обикновено се проявява при хора, които нямат фамилна анамнеза за това заболяване.
 Диагнозата се поставя чрез физикален преглед, извършване на рентгенологични изследвания и изследване на тъканни проби под микроскоп (хистология).
Диагнозата се поставя чрез физикален преглед, извършване на рентгенологични изследвания и изследване на тъканни проби под микроскоп (хистология).
На рентгеновото изображение се виждат:
- Скъсени ребра без фрактури
- Скъсени и разширени дълги кости
- Издължена фибула (малък пищял)
- Неправилна форма на таза
- Непълно вкостяване на някои кости
Пренатално диагностициране на хондрогенезис имперфекта също е възможно. То се извършва чрез ултразвуково изследване след 14-15-та гестационна седмица на бременността.
Лечение срещу заболяването няма. Терапията е насочена срещу съпътстващите симптоми и включва палиативни грижи, при които лекарите се опитват да намалят или сведат до минимум болката, стреса и дискомфорта, свързани с разстройството. Генетичното консултиране се препоръчва за семейства със засегнато дете. Психосоциалната подкрепа за цялото семейство също е от съществено значение.
-
Хипохондроплазия
Хипохрондроплазията представлява генетично заболяване, характеризиращо се с малък ръст и непропорционално къси ръце, крака, длани и ходила. Изоставането в ръста често не се разпознава до ранното детство или в някои случаи до средата на пубертета.
Хипохондроплазията засяга мъжете и жените в сравнително равен процент. Данни за заболяването са регистрирани за пръв път през 1913 година, а от тогава са записани над 100 случая в медицинската литература, включително спорадични случаи. Предполага се, че хипохондроплазията се среща приблизително дванадесет пъти по-рядко от ахондроплазията.
При децата с хипохондроплазия се появява и изкривяване на краката по време на ранното детство, но често се подобрява спонтанно с напредване на възрастта. Някои засегнати лица също могат да имат необичайно голяма глава (макроцефалия), изпъкнало и голямо чело и/или други физически аномалии, свързани с разстройството. Освен това, при около 10% от случаите може да има леко умствено изоставане.
Понякога хипохондроплазията се появява на случаен принцип по неизвестни причини (спорадично) без известна фамилна анамнеза, но в повечето случаи заболяването се предава от родителите на децата по автозомно-доминантен модел.
Най-честите симптоми на хипохондроплазията са:
- Анормално нисък ръст
- Скъсени крайници
- Макроцефалия
- Брахидактилия (необичайно къси пръсти)
- Аномалии на костите
При хората с хипохондроплазия, скъсяването на крайниците може да бъде относително леко или умерено. По време на ранното детство, обикновено се появява изкривяване в костите на краката, което се получава поради повишаване в теглото на децата, съчетано с неправилно развитие на костите. Интересното при това състояние е, че то често се подобрява по-късно по време на развитието.
Много от засегнатите лица също имат ограничено движение в лакътната става. При засегнатите възрастни ограничението на движенията може да прерасне до болка и дискомфорт в ставата, която да обхване и долната част на гърба. Приблизително една трета от пациентите страдат и от лордоза.
Хипохондроплазията се появява случайно по неизвестни причини (спорадично), без видима фамилна анамнеза за заболяването. Според изследователите такива случаи обикновено представляват нови генетични мутации, които могат да бъдат предадени по автозомно-доминантен модел.
 Докладвани са и фамилни случаи на нарушението. При тях разстройството има автозомно-доминантен характер на унаследяване, което означава, че едно копие на мутиралия ген във всяка клетка е достатъчно, за да предизвика аномалията.
Докладвани са и фамилни случаи на нарушението. При тях разстройството има автозомно-доминантен характер на унаследяване, което означава, че едно копие на мутиралия ген във всяка клетка е достатъчно, за да предизвика аномалията.
Рискът от предаване на разстройството от засегнатия родител на потомството е 50 процента за всяка бременност, независимо от пола на детето. Рискът е еднакъв за всяка бременност.
Изследователите посочват, че хипохондроплазията често се причинява от специфични мутации на ген, известен като "рецептор-3 на фибробластния растежен фактор" (FGFR3).
Изследователите също така са открили, че различни мутации на същия FGFR3 ген могат да причинят и ахондроплазия. Ахондроплазията, разбира се, се проявява в по-тежка форма, както беше описано по-горе.
Съществуват и данни след извършване на редица генетични анализи, че при някои индивиди с хипохондроплазия не се наблюдават мутации в FGFR3 гена. В такива случаи изследователите предполагат, че разстройството може да бъде резултат от мутации на други гени, които отново са свързани с FGFR3 гена.
Диагностиката се извършва с помощта на образно-диагностични и физикални методи. Както бе отбелязано по-рано, при  индивиди с хипохондроплазия, аномалията в ръста често може да не се прояви до първите няколко години от живота или до зряла възраст.
индивиди с хипохондроплазия, аномалията в ръста често може да не се прояви до първите няколко години от живота или до зряла възраст.
Лечението на хипохондроплазията обикновено е насочено срещу специфичните симптоми и се основава на хирургическа интервенция или физикална терапия. Генетично консултиране също се препоръчва за засегнатите лица и техните семейства. При бременни жени с хипохондроплазия често се изисква цезарово сечение за раждане.
Ранната намеса може да бъде решаваща, за да се гарантира, че засегнатите деца достигат своя потенциал за развитие. Допълнителни грижи, които могат да бъдат полезни, включват:
- Специализирано обучение на засегнатите индивиди
- Физиотерапия
- Професионална терапия
- Медицински, социални или професионални услуги
-
Нисък ръст (джудже)
Към групата на скелетните дисплазии се включва и ниският ръст (джудже).
Това заболяване се дължи на генетична аномалия или се разглежда като вторично, причинено от анормална функция на орган или система. За нисък ръст (джудже) обикновено се смята височина от 147 сантиметра или по-малко. Средната височина сред хората джуджета е 122 см.
Някои хора предпочитат понятието "нисък ръст" или "малки хора", пред "джудже". Заболяването не включва фамилния нисък ръст - ръст, който е в нормалните граници и с нормално развитие на костите.
Много различни медицински заболявания причиняват патологично нисък ръст.
По принцип това състояние може да се раздели на две големи категории:
- Непропорционален нисък ръст
- Пропорционален нисък ръст

- Непропорционален нисък ръст
Характеризира се с непропорционални размери на тялото. Някои части на тялото са абнормно малки, други са със среден или над средния размер. Нарушенията, при които се наблюдава непропорционален нисък ръст, се дължат на възпрепятстване на развитието на костите.
Повечето хора с аномалията имат нарушения, които причиняват непропорционално ниския ръст. Обикновено при непропорционалния нисък ръст пациентите имат нормален по размери гръден кош и торс, но със скъсени крайници или драстично намален торс с нормални по размер крайници. Често при този вид нисък ръст главата е непропорционално голяма в сравнение с тялото.
В повечето случаи на хора с непропорционален нисък ръст (джудже) не се наблюдават промени в интелектуалния капацитет. Редките изключения, при които се наблюдава умствен дефицит, обикновено са резултат от вторичен фактор, като например натрупване на излишна течност в черепната кутия (хидроцефалия).
капацитет. Редките изключения, при които се наблюдава умствен дефицит, обикновено са резултат от вторичен фактор, като например натрупване на излишна течност в черепната кутия (хидроцефалия).
Най-честата причина за ниския ръст е ахондроплазията. Това разстройство обикновено води до следното:
- Торс със среден размер
- Скъсени ръце и крака
- Скъсени пръсти на крайниците
- Ограничение в движенията на лакътните стави
- Непропорционално голяма глава, с изпъкнало чело
- Височина от около 122 см.
Друга причина за непропорционалния нисък ръст е рядкото заболяване, наречено спондилоепифизеална дисплазия конгенита. Симптомите могат да включват:
- Много намален по размери торс
- Скъсен врат
- Къси ръце и крака
- Широки, заоблен гръден кош
- Цепнатина на небцето
- Деформации на бедрата
- Прогресивно изкривяване на горната част на гръбначния стълб
- Проблеми със слуха
- Артрит и проблеми със ставното движение
- Височина, варираща от 91 см. до 122 см.
- Пропорционален нисък ръст

Характеризира се с пропорционално малко тяло, при което всички негови части са с намалени размери в една и съща степен и изглеждат пропорционални на тялото.
Пропорционалният нисък ръст (джудже) се дължи на заболявания, проявяващи симптомите си веднага след раждането или появяващи се в ранна детска възраст, които ограничават цялостния растеж и развитие на костите.
Главата, торса и крайниците са с анормално малки размери, но са пропорционални един на друг. Тъй като тези нарушения засягат цялостния растеж, много от тях водят до проблеми и забавяне в развитието на една или повече системи на тялото.
Дефицитът на хормона на растежа е сравнително често срещана причина за заболяването. Това се случва, когато хипофизната жлеза не успее да произведе достатъчно количество хормон на растежа, което е от съществено значение за нормалния детски растеж.
Признаците включват:
- Намален ръст
- Забавен растеж и развитие
- Забавено или никакво сексуално развитие през юношеските години
Причините за ниския ръст (джуджета) са често генетични или органни заболявания, но има и случаи, в които факторите са неизвестни. Повечето случаи на нисък ръст произтичат от случайна генетична мутация в гените на бащата или майката, като се предават в потомството, но мутациите могат да възникнат и спорадично в засегнатия индивид (de novo).
Както вече беше споменато ахондроплазията е една от честите причини за ниския ръст. Около 80% от пациентите с ахондроплазия имат родители с нормална височина, което означава, че мутацията възниква спорадично. Когато ахондроплазията се унаследява, това се случва по автозомно-доминантен модел.
Съществува 25% вероятност едно дете, родено от двойка, в която и двамата родители страдат от заболяването, да се роди без промени в растежа.
Синдромът на Търнър е едно от другите заболявания, които причиняват ниския ръст. Синдромът засяга само момичета и жени и се дължи на липсваща  Х-хромозома. Едно момиче със синдром на Търнър има само едно напълно функциониращо копие на женската полова хромозома, а не две, както е нормално.
Х-хромозома. Едно момиче със синдром на Търнър има само едно напълно функциониращо копие на женската полова хромозома, а не две, както е нормално.
Усложненията при ниския ръст могат да се различават значително, но някои усложнения са често срещани при редица заболявания. Характерните аномалии на черепа, гръбначния стълб и крайниците водят до някои общи проблеми:
- Забавяне в развитието на моторни умения, като например седене, пълзене и ходене
- Повтарящи се инфекции на ушите и риск от загуба на слуха
- Сънна апнея
- Излишната течност около мозъка (хидроцефалия)
- Проблеми с дишането
- Стеноза (стеснение) на гръбначния канал
- Артрит
- Недоразвити органи
Някои форми на ниския ръст (джудже) са очевидни още по време на вътреутробното развитие, при раждането или в ранна детска възраст. Те могат да бъдат диагностицирани чрез образно-диагностични или кръвни изследвания, както и след оглед от страна на лекар.
Ако малформациите се дължат на вродени дисплазии, диагнозата може да бъде потвърдена и чрез пренатална диагностика. В някои случаи изследването се прави, ако родителите страдат от други специфични заболявания на опорно-двигателния апарат, които се предават в семейството.
 Обикновено част от медицинския преглед на пациента включва измерването на височината, теглото и обиколката на главата.
Обикновено част от медицинския преглед на пациента включва измерването на височината, теглото и обиколката на главата.
Това е важно за идентифицирането на необичайно забавен растеж или непропорционално голяма глава.
Извършват се и рентгенологични и ядрено-магнитни изследвания, тъй като някои аномалии на черепа и скелета могат да помогнат за уточняването на диагнозата. Тези методи също могат да разкрият забавено вкостяване и растеж на костите, какъвто е случаят с дефицита на растежен хормон. Ядрено-магнитния резонанс (ЯМР) може да разкрие аномалии на хипофизната жлеза или хипоталамуса, като и двете играят роля в хормоналната функция.
Потенциални признаци и симптоми, които се търсят при съмнение за нисък ръст (джудже) са:
- Непропорционално голяма глава
- Късното развитие на някои двигателни умения, като например седене или ходене
- Проблеми с дишането
- Изкривяване на гръбначния стълб
- Ставни проблеми
- Болка в гърба или изтръпване в краката
Лечението при това заболяване има за цел да подобри възможно най-много функционирането и независимостта на засегнатите индивиди. В повечето случаи не се стреми увеличаване на ръста, а да се коригират проблемите и усложненията от малформациите.
Хирургическите интервенции, които могат да коригират проблемите при хора с нисък ръст (джудже), включват:
- Коригиране на посоката, в която костите растат
- Стабилизиране и коригиране на формата на гръбначния стълб
- Облекчаване на натиска върху гръбначния мозък
- Поставяне на шънт за отстраняване на излишната течност около мозъка (хидроцефалия).
- Трахеотомия за подобряване на дишането
- Премахване на сливиците за подобряване на дихателните проблеми
За хора с нисък ръст, дължащ се на дефицит на растежен хормон, лечението със синтетична форма на хормона може да увеличи в известна степен ръста. В повечето случаи пациентите от детска възраст получават ежедневни дози от хормона в продължение на няколко години, докато достигнат нормална височина.
повечето случаи пациентите от детска възраст получават ежедневни дози от хормона в продължение на няколко години, докато достигнат нормална височина.
Лечението може да продължи и през юношеските години, за да се осигури нормално съзряване по време на пубертета. Някои хора може да се нуждаят от терапия през целия живот. Лечението може да бъде допълнено и с други хормони.
Лечението на момичета със синдром на Търнър също изисква естроген и свързаната със заболяването хормонална терапия, за да навлязат в периода на пубертета. Естрогенната заместителна терапия обикновено продължава през целия живот.
За съжаление прилагането на растежен хормон при деца с ахондроплазия не увеличава ръста им.
-
Остеосклерозис конгенита (вродена остеосклероза)
Вродената остеосклероза е последното заболяване от групата на ахондроплазиите. Тя често се свързва с други заболявания като ахондроплазия, остеопетроза, остеопойкилоза и др. Характеризира се с увеличаване на костното вещество и абнормно повишаване на костната плътност (втвърдяване). Това води до значително увеличаване на риска от фрактури, поради намалената еластичност на костта.
Причината за вродената остеосклероза все още не е известна, както и начина на възникване или предаване. Предполага се, че се случва в следствие на мутация в ген, отговорен за изграждането или вкостяването на костите.
Характерната находка при рентгенологично изследване е прекомерно нарастване на ендооста (разделя костната тъкан от костния мозък), без да се наблюдава промяна в дължината на костта.
Обикновено се засягат всички кости в организма, но при някои промените са по-изявени.
 Когато вродената остеосклероза се развие напълно се наблюдава еднородно (хомогенно) задебеляване на засегнатите кости, което се проявява с по-светли участъци от костта на рентгенография.
Когато вродената остеосклероза се развие напълно се наблюдава еднородно (хомогенно) задебеляване на засегнатите кости, което се проявява с по-светли участъци от костта на рентгенография.
Тези промени се наблюдават най-често в телата на прешлените, тазовите кости и основата на черепа.
Пациентите с това заболяване нямат конкретни оплаквания от вродената остеосклероза, като по-скоро се наблюдават симптоми на свързаните с остеосклерозата заболявания.
Диагностиката както беше споменато се извършва чрез рентгенологично изследване на частите на тялото.
Лечение за това заболяване няма. Терапията по-скоро е насочена към придружаващите заболявания.
Симптоми и признаци при Ахондроплазия МКБ Q77.4
- Ментална ретардация
- Гърчове
- Нисък ръст
- Цепка на небцето
- Намален мускулен тонус
- Проблеми в развитието
Библиография
https://ghr.nlm.nih.gov/condition/achondroplasia
https://www.healthline.com/health/achondroplasia
https://www.stanfordchildrens.org/en/topic/default?id=achondroplasia-90-P01938
https://rarediseases.org/rare-diseases/achondroplasia/
https://emedicine.medscape.com/article/1258401-treatment#showall
https://rarediseases.info.nih.gov/diseases/914/chondrodysplasia-blomstrand-type
https://www.dovemed.com/diseases-conditions/blomstrand-lethal-chondrodysplasia/
https://link.springer.com/referenceworkentry/10.1007%2F978-3-540-29676-8_236
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3403254/
https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/chondrogenesis
https://ghr.nlm.nih.gov/condition/hypochondroplasia
https://rarediseases.info.nih.gov/diseases/6724/hypochondroplasia
https://rarediseases.org/rare-diseases/hypochondroplasia/
https://www.webmd.com/children/dwarfism-causes-treatments#3-5
https://www.mayoclinic.org/diseases-conditions/dwarfism/symptoms-causes/syc-20371969
https://www.medicalnewstoday.com/articles/320286.php
https://pubs.rsna.org/doi/full/10.1148/rg.317115093
https://www.jpeds.com/article/S0022-3476(33)80094-1/abstract
Коментари към Ахондроплазия МКБ Q77.4