Ахондрогенеза МКБ Q77.0
Група от редки скелетни дисплазии (ненормално развитие на тъкан или орган), характеризиращи се с изключително скъсяване на ръцете и краката в сравнение с гръдния кош, необичайно развитие на ребрата, прешлените и други скелетни аномалии се обединяват под термина ахондрогенеза. Здравните проблеми, свързани с тези състояния, са животозастрашаващи и най-засегнатите бебета често са мъртвородени или умират скоро след раждането, поради тежката дихателна недостатъчност.
Всички видове ахондрогенеза представляват много тежки скелетни дисплазии, които обикновено се откриват чрез пренатална ултразвукова диагностика още през 14-17 седмица от гестационния период.
Терминът ахондрогенеза е използван за първи път в медицинската литература през 1952 г. от италианския патолог Марко Фраккаро. Ахондрогенезата произлиза от гръцки език и означава "не произвежда хрущял".
Това заболяване принадлежи към група скелетни дисплазии (наричани още остеохондродисплазии) - широк термин за група от заболявания (около 450 клинични диагнози), характеризиращи се с ненормален растеж или развитие на хрущял и кост.
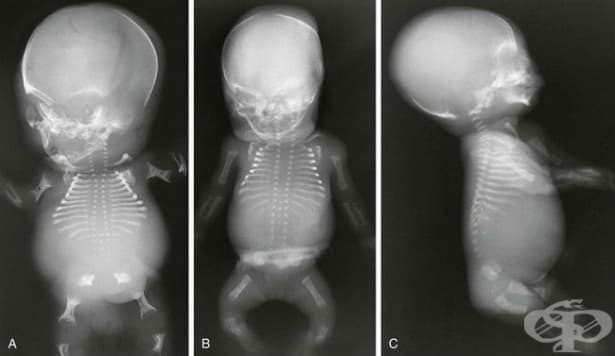
 Ахондрогенезата се характеризира с преждевременно раждане, прекомерно натрупване на течност в организма (hydrops fetalis) и глава, която може да има анормална форма и да не бъде напълно вкостена. Главата може да изглежда непропорционално голяма, поради малкия размер на тялото. В допълнение, засегнатите индивиди имат изключително къси крайници и ребра, къс врат, плоски прешлени и много други неправилно развити кости на скелета.
Ахондрогенезата се характеризира с преждевременно раждане, прекомерно натрупване на течност в организма (hydrops fetalis) и глава, която може да има анормална форма и да не бъде напълно вкостена. Главата може да изглежда непропорционално голяма, поради малкия размер на тялото. В допълнение, засегнатите индивиди имат изключително къси крайници и ребра, къс врат, плоски прешлени и много други неправилно развити кости на скелета.
При кърмачета, родени с това заболяване, коремът е изпъкнал, а гръдният кош е малък.
Други аномалии са непълно затваряне на устната кухина (цепнатина на небцето), дефекти на роговицата и деформации на ушите. Разстройството е животозастрашаващо преди раждането или малко след раждането, обикновено поради недоразвит гръден кош и недоразвити бели дробове.
Всеки тип ахондрогенеза се причинява от мутация в специфичен ген. Гените осигуряват инструкции за създаване на протеини, които играят важна роля в много функции на тялото. Когато възникне мутация на ген, създаденият протеин може да бъде дефектен, неефективен или отсъстващ. В зависимост от функциите на конкретния белтък, това може да засегне много системи на тялото.
Описани са три основни типа ахондрогенеза, означени като:
- тип 1 А (Хюстън-Харис тип)
- тип 1 В (Паренти-Фраккаро тип)
- тип 2 (Лангер-Салдино тип)
Типовете се отличават с техните характерни признаци и симптоми, модел на унаследяване и генетична мутация. Въпреки това типове 1 А и 1 В често са трудни за разграничаване без генетично изследване.
Ахондрогенезата тип 1 A и тип 1 B са много редки заболявания и разпространението им е неизвестно. Ахондрогенезата тип 2 възниква при приблизително 1/40,000 - 1/60,000 новородени.
Генните мутации, които причиняват ахондрогенеза тип 1 A и тип 1 B, се унаследяват по автозомно-рецесивен модел, което означава, че индивидът трябва да унаследи две променени копия на мутиралия ген във всяка клетка, за да бъде засегнат.
Родителите на индивид с автозомно-рецесивно заболяване носят по едно копие на мутиралия ген, но те обикновено не показват признаци и симптоми на синдрома. Ако дадено лице получи един нормален ген и един ген за болестта, лицето е носител на болестта, но обикновено няма да показва симптоми.

Рискът за двама родители-носители да имат засегнато дете е 25% при всяка бременност. Рискът да имат дете, което е носител като тях самите, е 50%
цепнатина на небцето при всяка бременност. Възможността детето да получи нормални гени от двамата родители е 25%. Рискът е еднакъв за мъжете и жените.
Родители, които са близки роднини, имат по-голям шанс да носят същия анормален ген. Това увеличава риска да има деца с рядко рецесивно генетично заболяване в семейство с близкородствен брак.
Тип 1-А се причинява от мутации в гена TRIP11.
Тип 1 В се причинява от мутации в гена SLC26A2.
Тип 2 ахондрогенезата се причинява от нови (de novo) мутации в COL2A1 гена.
Ахондрогенезата тип 1 А, която също се нарича тип Houston-Harris, е най-слабо проучена от трите форми. Засегнатите деца имат изключително къси крайници, тесен гръден кош, къси ребра, които лесно се чупят, и липса на нормално образуване на кости (осификация) в черепа, гръбначния стълб и таза. Поради малкия размер на гръдния кош белите дробове не се развиват напълно и това е основната причина за смърт при новородените.
Ахондрогенеза тип 1 В, известен също като тип Parenti-Fraccaro, се характеризира с изключително къси крайници, тесен гръден кош и изпъкнал, заоблен корем. Пръстите и краката са къси, а ходилата могат да се обърнат навътре и нагоре. Засегнатите бебета често имат пъпна или ингвинална херния.
Ахондрогенеза тип 2, която се нарича още тип Langer-Saldino, имат къси ръце и крака, тесен гръден кош с къси ребра и недоразвити бели дробове. Това състояние е свързано и с липсата на осификация в гръбначния стълб и таза. Отличителните черти на лицето включват изпъкнало чело, малка брадичка и цепнатина на небцето. Коремът е подут, а засегнатите бебета често страдат от състояние, наречено hydrops fetalis, при което излишната течност се натрупва в тялото преди раждането.

Диагнозата се поставя чрез физикален преглед, извършване на рентгенологични изследвания и изследване на тъканни проби под микроскоп (хистология). Молекулярни генетични тестове за мутации в SLC26A2 гена могат да бъдат използвани за потвърждаване на диагнозата ахондрогенеза тип 1 В.
Пренаталното диагностициране на ахондрогенезата чрез ултразвуково изследване е възможно след 14-15-та гестационна седмица на бременността.
Пренатална диагностика чрез вземане на хорионни вили (10-12-та гестационна седмица) или амниоцентеза (15-18-та гестационна седмица) е възможна, ако специфичните генни мутации са идентифицирани в член на семейството.
Лечение срещу ахондрогенезата няма. Терапията е насочена срещу симптомите на заболяването и включва палиативни грижи, при които лекарите се опитват да намалят или сведат до минимум болката, стреса и специфичните симптоми, свързани с разстройството. Генетичното консултиране се препоръчва за семейства със засегнато дете. Психосоциалната подкрепа за цялото семейство също е от съществено значение.
Друго заболяване на костната система, свързано с ахондрогенезата е хипохондрогенезата. Тя  представлява тежко генетично заболяване, характеризиращо се с малформации в костния растеж. При пациентите се наблюдават късо тяло и крайници, както и необичайно образуване на костите от състава на гръбначния стълб и таза.
представлява тежко генетично заболяване, характеризиращо се с малформации в костния растеж. При пациентите се наблюдават късо тяло и крайници, както и необичайно образуване на костите от състава на гръбначния стълб и таза.
Хипохондрогенезата е сходна с ахондрогенезата тип 2, въпреки че промените в гръбначния стълб, наблюдавани при хипохондрогенезата, са по-леки.
Засегнатите деца имат къси ръце и крака, малък гръден кош с къси ребра и недоразвити бели дробове. Костите в черепа се развиват нормално, но костите на гръбначния стълб и таза не се вкостяват правилно. Лицето изглежда плоско и овално, с раздалечени очи, малка брадичка и в някои случаи непълно затваряне на устната кухина (цепнатина на небцето).
Хората с хипохондрогенеза имат подут корем и може да имат hydrops fetalis.
В резултат на тези сериозни здравословни проблеми някои засегнати фетуси не оцеляват до раждането. Деца, родени с хипохондрогенеза обикновено умират при раждането или малко след това от дихателна недостатъчност.
Подобно на ахондрогенезата се наблюдава засягане в средно 1 на 40,000 до 60,000 новородени.
Хипохондрогенезата е едно от най-тежките състояния в спектъра на нарушения, причинени от мутации в гена COL2A1. Този ген осигурява инструкции за създаване на протеин, който образува колаген тип II. Този тип колаген се открива предимно в стъкловидното тяло на очната ябълка и в хрущяла.
Хрущялът представлява здрава, гъвкава тъкан, която съставя голяма част от скелета по време на ранното му развитие. Повечето хрущяли по-късно се превръщат в кости, с изключение на хрущяла, продължаващ да покрива и защитава краищата на костите и присъстващ в носа и външните уши.
Колаген тип II е от съществено значение за нормалното развитие на костите и другите съединителни тъкани. Мутациите в гена COL2A1 пречат на сглобяването на молекули на колаген тип II, което предотвратява правилното развитие на костите и другите съединителни тъкани.
Хипохондрогенезата се счита за автозомно-доминантно заболяване, защото едно копие на променения ген във всяка клетка е достатъчно, за да причини това състояние. То се причинява от нови мутации в гена COL2A1 и се проявява при хора, които нямат история на разстройството в семейството си.
Това заболяване не се предава в следващото поколение, защото засегнатите лица не живеят достатъчно дълго, за да имат деца.
 Диагнозата на хипохондрогенезата може да се направи пренатално чрез ехографско изследване или различни ДНК тестове. За потвърждаване на диагнозата се използват рентгенологични изследвания, биопсии и др. Консултация с експерти в областта на скелетната дисплазия също може да бъде полезна.
Диагнозата на хипохондрогенезата може да се направи пренатално чрез ехографско изследване или различни ДНК тестове. За потвърждаване на диагнозата се използват рентгенологични изследвания, биопсии и др. Консултация с експерти в областта на скелетната дисплазия също може да бъде полезна.
Няма лечение за хипохондрогенезата. Ако диагнозата е направена непосредствено преди раждането, родителите може да пожелаят да се срещнат с неонатолог, за да обсъдят вариантите пред себе си.
Ако състоянието се открие по време на бременността, пациентите имат възможност да прекратят бременността въз основа високата смъртност при състоянието.
Това е много лично решение, което трябва да бъде взето след сериозно консултиране относно естеството и резултата от тази диагноза.
Симптоми и признаци при Ахондрогенеза МКБ Q77.0
- Затруднено дишане
- Нисък ръст
- Симптоми на челюстта
- Цепка на небцето
- Симптоми от пръстите
- Симптоми на шията
Библиография
https://rarediseases.info.nih.gov/diseases/2882/achondrogenesis
https://www.healthline.com/health/achondrogenesis
https://emedicine.medscape.com/article/941176-overview
https://www.encyclopedia.com/science/encyclopedias-almanacs-transcripts-and-maps/hypochondrogenesis
Коментари към Ахондрогенеза МКБ Q77.0