Друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб МКБ Q77.8
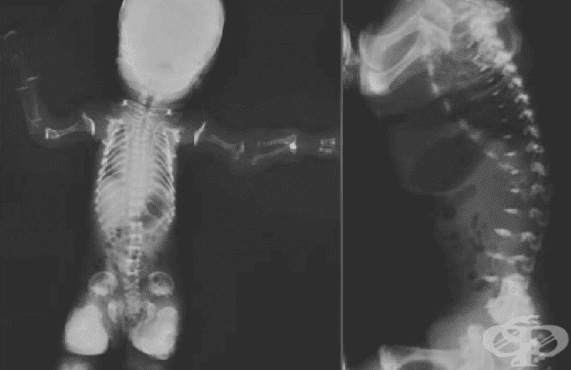
Остеохондродисплазия или скелетна дисплазия е общ термин за нарушение в развитието (дисплазия) на костите ("остео") и хрущяла ("хондро").
Остеохондродисплазиите са редки заболявания. Около 1 на 5 000 бебета се раждат с някакъв тип скелетна дисплазия. Остеохондродисплазиите могат да доведат до значително функционално ограничение, дори и смърт.
Остеохондродисплазиите са много видове и могат да се припокриват в някои клинични аспекти. Следователно, рентгенологичното изследване е абсолютно необходимо за установяване на точна диагноза. Диагностиката чрез ядрено-магнитен резонанс (ЯМР) може да осигури допълнителна диагностична информация и да насочи специалистите към точната диагноза. Ранната диагноза и своевременното лечение на скелетната дисплазия са важни за борбата с функционалното влошаване.
Заболяванията, включени в тази група са:
- Метатропна дисплазия (Metatropic Dysplasia)
- Дисхондростеозата на Лери-Вайл (Leri-Weill dyschondrosteosis)
- Мезомелична дисплазия на Лангер (Langer mesomelic dysplasia)
- Акромезомелична дисплазия (Acromesomelic Dysplasia)
- Дисплазия на Астли-Кендал (Astley-Kendall syndrome)
- Кампомеличен синдром (Campomelic Syndrome)
- Хондродисплазия с охлювоподобен таз (Schneckenbecken dysplasia)
- Синдром на Уолкот-Ралисон (Wolcott-Rallison syndrome)
Метатропна дисплазия (Metatropic Dysplasia)
Метатропната дисплазия представлява рядко генетично заболяване, характеризиращо се с изключително нисък ръст, къси ръце и крака. Други особености на това разстройство са:
- стеснен гръден кош
- къси ребра
- кифоза или сколиоза, която се развива хората с нисък ръст (джуджета)
 Метатропната дисплазия се характеризира с анормално развитие на скелета. Пациентите с това разстройство обикновено имат скъсени ребра, къси деформирани ръце и крака, кифосколиоза (необичайно изкривяване на гръбначния стълб) и изключително нисък ръст. Издължен и тесен гръден кош, задебелени ставни повърхности с ограничена подвижност на коленете и бедрата, както и необичайно удължаване на ставите на пръстите също са характерни особености.
Метатропната дисплазия се характеризира с анормално развитие на скелета. Пациентите с това разстройство обикновено имат скъсени ребра, къси деформирани ръце и крака, кифосколиоза (необичайно изкривяване на гръбначния стълб) и изключително нисък ръст. Издължен и тесен гръден кош, задебелени ставни повърхности с ограничена подвижност на коленете и бедрата, както и необичайно удължаване на ставите на пръстите също са характерни особености.
Торсът е издължен, а това е ранна характеристика на метатропната дисплазия. Рентгенологичното изследване показва недостатъчност в растежа на гръбначния стълб с аномалии в прешлените и промени в костите на ръцете и краката, както и в ставите.
Метатропната дисплазия може да се наследи както по автозомно-доминантен, така и по автозомно-рецесивен модел.
Човешките белези и характерни черти, включително и генетичните заболявания, са продукт на взаимодействието на два гена, единият получен от бащата, а другият от майката.
При заболяванията, които се унаследяват доминантно е нужно едно единствено копие на мутиралия ген (получено от майката или бащата), за да се развие болестта. Рискът от предаване на аномалията от родител на дете е 50 процента за всяка бременност, независимо от пола на детето.
При заболявания, които се предават рецесивно болестното състояние не се проявява, освен ако човек не наследи мутирал ген едновременно и от двамата родители. Ако дадено лице получи един нормален ген и един мутирал ген, лицето ще бъде здрав носител на болестта и няма да показва симптоми.
Рискът от предаване и изявяване на болестта при рецесивните заболявания е 25%, докато 50% от децата на такива родители рискуват да бъдат здрави носители, а 25% е шансът да получат напълно нормални и непроменени гени. Рискът е еднакъв за всяка бременност.
Метатропната дисплазия е много рядко заболяване, което засяга мъже и жени в еднаква степен.
Симптомите на следните заболявания могат да бъдат подобни на тези на метатропната дисплазия:
Синдром на Книест (Kniest Syndrome)
Синдром на Моркьо (Morquio Syndrome)
Синдромът на Книест е рядък тип нисък ръст (джудже), характеризиращ се с необичайно къси ръце и крака, кръгло лице, подуване и скованост на ставите и пръстите. Възможно е също така да се наблюдават аномалии на небцето, сколиоза, зрителни проблеми и проблеми със слуха.
Синдромът на Моркьо е метаболитно нарушение, характеризиращо се с натрупване на кератан сулфат. В резултат на този дефект могат да възникнат костни отклонения на черепа, гръдния кош, ръцете, коленете и гръбначния стълб. Интелектът обикновено не е засегнат. Костните аномалии на гръбначния стълб могат да доведат до компресия (притискане) на гръбначния мозък. Също така може да има увеличение на черния дроб, изкривяване на гръбначния стълб, аномалии на сърцето, както и загуба на слуха.
Диагностиката на метатропната дисплазия се основава на клинични и образно-диагностични изследвания. Находките при рентгенография включват къси диафизи (средна част на костите) с широки метафизи (частта от костта между епифизата и диафизата), платиспондилия (сплескване на телата на гръбначните прешлени), калцификация на хрущяли, аномалии на таза и др. Интересно при това състояние е, че рентгенологичните прояви се изменят с напредването на възрастта. Молекулярно-генетичното изследване може да идентифицира мутация в TRPV4 гена, което също потвърждава диагнозата.
 Пренатална диагностика е възможна чрез извършване на изследване с компютърен томограф или ултразвук. Генетично консултиране също е възможно.
Пренатална диагностика е възможна чрез извършване на изследване с компютърен томограф или ултразвук. Генетично консултиране също е възможно.
Лечението на вродената аномалия е насочено към забавяне на прогресията на скелетната деформация, както и към поддържане на нормална белодробна функция. Стандартна практика е прилагането на ортопедични шини или колани в периода на растеж и развитие.
Обикновено се предпочита консервативен подход, но хирургичните процедури могат да бъдат много успешни в някои случаи за превенция на деформациите. При тежки случаи с дихателно увреждане може да е необходима трахеостома и дългосрочна вентилационна подкрепа.
Дисхондростеозата на Лери-Вайл (Leri-Weill dyschondrosteosis)
Друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб е дисхондростеозата на Лери-Вайл.
Дисхондростеозата на Лери-Вайл (Leri-Weill dyschondrosteosis) е рядко генетично заболяване, характеризиращо се с необичайно скъсяване на предмишниците и подбедриците, деформация на китката (Маделунг (Madelung) деформация на предмишницата и ръката) и свързания със заболяването нисък ръст.
Могат да се появят и допълнителни симптоми. Тези допълнителни симптоми и тяхната тежест могат да се различават значително от един човек на друг, дори между членовете на едно и също семейство. Интелектуалните способности не са засегнати.
 Дисхондростеозата на Лери-Вайл е рядко заболяване, което може да засегне както мъжете, така и жените. Повече случаи на това нарушение са съобщени при жените, като съотношението е 4:1 в тяхна полза.
Дисхондростеозата на Лери-Вайл е рядко заболяване, което може да засегне както мъжете, така и жените. Повече случаи на това нарушение са съобщени при жените, като съотношението е 4:1 в тяхна полза.
Преобладаването на вродената аномалия е неизвестно, но често се определя като между 1 на 1000-2000 души от популацията. Въпреки това много засегнати индивиди могат да бъдат неправилно диагностицирани, което затруднява определянето на точната честота на дисхондростеозата на Лери-Вайл в световното население.
Дисхондростеозата на Лери-Вайл се причинява от мутация в SHOX гена или неговите регулаторни елементи, разположени в половите хромозоми.
Дисхондростеозата на Лери-Вайл е описана за първи път в медицинската литература през 1929 г. от лекарите Leri и Weill. Разстройството представлява скелетна дисплазия и се асоциира с мутации в SHOX гена или неговите енхансери (елементи в гените, които подпомагат генетичните процеси). Допълнителни нарушения в гените включват по-тежки промени в скелетната структура, като например мезомеличната дисплазия на Лангер. При нея засегнатите лица обикновено имат голямо скъсяване на дългите кости в ръцете и краката (мезомелия), в резултат на което хората с мезомелична дисплазия на Лангер имат много нисък ръст.
Специфичните признаци и симптоми, свързани с дисхондростеозата на Лери-Вайл, могат да варират значително от човек на човек. Като цяло жените са с по-сериозни малформации от мъжете.
Класическите симптоми на разстройството са:
- мезомелично съкращаване на крайниците
- нисък ръст
- деформация на Маделунг
При някои хора не се развива деформация на Маделунг и/или могат да имат нормална височина.
Мезомеличното скъсяване на крайниците предизвиква ненормално съкращаване на средната част на ръцете и краката. Това означава, че предмишниците и подбедриците са непропорционално по-къси от горната част на ръцете и краката. Следователно, ръцете и краката са непропорционални по размер на останалите части от тялото. Понякога големия пищял (тибията) и лъчевата и лакътната кост (радиус и улна) могат да бъдат патологично извити.
По-рядко може да се наблюдава болка в китката, коляното или глезена. Мезомелията обикновено се проявява за пръв път в училищна възраст, а симптомите може да се задълбочат с напредване на възрастта. При дисхондростеозата на Лери-Вайл степента на нисък ръст може да варира значително от един човек на друг. Често пациентите са с леко намалена височина, но има и случаи на драстично намален ръст.
Засегнатите лица също могат да имат аномалия на китката, известна като деформация на Маделунг, която става по-очевидна около пубертета. Тази деформация се характеризира с изкривяване и скъсяване на костите на предмишницата (радиус и улна), както и дислокация (разместване) на улната, което води до анормално отклонение или неправилна позиция и форма на китката. Обикновено се наблюдава двустранна деформация на Маделунг, тоест и двете китки са засегнати. Засегнатите индивиди могат да имат ограничен спектър от движения на китките и лактите и/или могат да изпитват болка, както и да имат видими промени във външния вид на китката.
Обикновено се наблюдава двустранна деформация на Маделунг, тоест и двете китки са засегнати. Засегнатите индивиди могат да имат ограничен спектър от движения на китките и лактите и/или могат да изпитват болка, както и да имат видими промени във външния вид на китката.
Деформацията на Маделунг на китката може да възникне и от травма или инфекция. В тези случаи тя често не е двустранна, т.е. не засяга двете китки. Деформация на Madelung може да възникне и като част от синдрома на Търнър (10% от случаите).
Допълнителните симптоми могат да включват:
- деформации на небцето
- къси и задебелени кости на ръката
- сколиоза
- хипертрофия на мускулите на подбедрицата
В повечето случаи дисхондростеозата на Лери-Вайл се причинява от мутации в или загуба (делеция) на SHOX гена или неговите регулаторни региони. Този ген осигурява инструкции за създаване на протеини, които играят важна роля в много функции на тялото. Когато възникне мутация, продуктите от синтеза на протеини може да бъдат дефектни, неефективни или отсъстващи. В зависимост от функциите на конкретния белтък, това може да засегне много органни системи на тялото.
 Генните промени, които причиняват дисхондростеозата на Лери-Вайл, се наследяват по автозомно- или псевдоавтозомно-доминантен механизъм. Псевдоавтозомно-доминантното наследяване е изключително рядко явление, което освен мутация в автозомите (всички 22 чифта хромозоми, без половите), включва и мутирали гени, намиращи се в някоя от двете полови хромозоми - X или Y.
Генните промени, които причиняват дисхондростеозата на Лери-Вайл, се наследяват по автозомно- или псевдоавтозомно-доминантен механизъм. Псевдоавтозомно-доминантното наследяване е изключително рядко явление, което освен мутация в автозомите (всички 22 чифта хромозоми, без половите), включва и мутирали гени, намиращи се в някоя от двете полови хромозоми - X или Y.
Мутирал ген, разположен на автозома може да бъде предаден на дете от мъжки или женски пол с еднаква вероятност. Това се нарича автозомно унаследяване.
Въпреки това, половите хромозоми (X и Y) не се предават равномерно, защото бащата предава своята X хромозома на дъщерите си, а Y хромозомата на синовете си. Това се нарича свързано с пола унаследяване.
Ключов аспект на свързаното с пола унаследяване е липсата на съвпадащи генни двойки между X и Y хромозомите. Съществува обаче явление, наречено кросингоувър, което представлява размяна на част от Х-хромозомата с Y-хромозомата. По този начин мутацията на SHOX гена може да бъде предадена от едната хромозома на другата и да се наблюдава псевдоавтозомно-доминантното унаследяване.
Диференциалната диагноза при дисхондростеозата на Лери-Вайл трябва да включва другите SHOX-свързани аномалии, както и свързаните с тях състояния като синдром на Търнър и др.
Диагностиката на дисхондростеозата на Лери-Вайл се основава на задълбочен клиничен преглед и характеристика на физикалните находки. Диагнозата в някои случаи се поставя трудно, защото някои симптоми може да не се проявят чак до пубертета. Рентгенологичното изследване, по-специално рентгенографията на китката, могат да разкрият характерни промени в засегнатите кости.
Молекулярно-генетичното изследване може да потвърди диагнозата в приблизително 70% от случаите.
 Лечението на заболяването е насочено към симпомите. Генетичното консултиране може да бъде от полза за засегнатите лица и техните семейства. Лечението с хормон на растежа е препоръчително за деца, които не са достигнали до пубертета. Според медицинската литература може да се постигне увеличаване в ръста от 7 до 10 сантиметра, при прилагане на хормона на растежа. Скелетните дефекти също не се влошават с лечението.
Лечението на заболяването е насочено към симпомите. Генетичното консултиране може да бъде от полза за засегнатите лица и техните семейства. Лечението с хормон на растежа е препоръчително за деца, които не са достигнали до пубертета. Според медицинската литература може да се постигне увеличаване в ръста от 7 до 10 сантиметра, при прилагане на хормона на растежа. Скелетните дефекти също не се влошават с лечението.
Деформацията на Маделунг не изисква терапия. Използването на ергономични устройства, предназначени да облекчат болката в китката, може да бъде от полза. Ако деформацията на Маделунг причинява болка или дискомфорт, дейностите, които предизвикват болката, трябва да бъдат ограничени. Някои хора могат да имат тежка форма на тази деформация и да се нуждаят от ортопедична операция за облекчаване на болката и подобряване на движенията.
Към групата на друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб се причисляват и други заболявания като:
- Мезомелична дисплазия на Лангер (Langer mesomelic dysplasia)
- Акромезомелична дисплазия (Acromesomelic Dysplasia)
- Дисплазия на Астли-Кендал (Astley-Kendall syndrome)
- Кампомеличен синдром (Campomelic Syndrome)
- Хондродисплазия с охлювоподобен таз (Schneckenbecken dysplasia)
- Синдром на Уолкот-Ралисон (Wolcott-Rallison syndrome)
- Мезомелична дисплазия на Лангер
Мезомеличната дисплазия на Лангер е много рядка форма на нисък ръст (джудже), която засяга мъже и жени в еднаква степен и се наследява по автозомно- или псевдоавтозомно-рецесивен механизъм. SHOX мутации на двете полови хромозоми, т.е. 2 мутации, причиняват този вид скелетна дисплазия.
еднаква степен и се наследява по автозомно- или псевдоавтозомно-рецесивен механизъм. SHOX мутации на двете полови хромозоми, т.е. 2 мутации, причиняват този вид скелетна дисплазия.
Хората с това разстройство обикновено са с много скъсен ръст (около 125 см за възрастни), дължащ се на къси, задебелени, извити кости на подбедрицата.
Недостатъчен растеж или развитие и патологично огъване на костите на предмишницата също присъства.
Други особености на това разстройство могат да бъдат:
- ограничено движение на лактите и предмишницата
- слабо развита долна челюст
- лордоза
Заболяването се причинява от мутация в SHOX гена, а диагностиката и лечението са като на дисхондростеозата на Лери-Вайл.
- Акромезомелична дисплазия
Акромезомеличната дисплазия е изключително рядко, наследено и прогресивно скелетно заболяване, което води до особена форма на нисък ръст с драстично скъсени крайници. Разстройството се характеризира с акромелия (скъсяване на костите на ръцете и ходилата) и мезомелия (скъсени предмишници и подбедрици). По този начин, ниският ръст на засегнатите индивиди е резултат от необичайно съкращаване на костите на подбедриците, като се наблюдава и необичайно скъсяване на предмишниците. Много характерни са късите ръце и късите пръсти. Тези белези са очевидни още през първите години от живота.
 Децата с акромезомелична дисплазия често имат нормално тегло при раждане. В повечето случаи, освен че имат необичайно къси и широки ръце и крака, засегнатите бебета често имат характерни аномалии на лицето, които се наблюдават при раждането.
Децата с акромезомелична дисплазия често имат нормално тегло при раждане. В повечето случаи, освен че имат необичайно къси и широки ръце и крака, засегнатите бебета често имат характерни аномалии на лицето, които се наблюдават при раждането.
Аномалиите на лицето включват:
- макроцефалия
- изпъкнало чело
- необичайно малък нос
През първите години от живота, когато предмишниците, подбедриците, дланите и стъпалата не нарастват пропорционално с останалата част от тялото, се проявяват първите симптоми на характерния нисък ръст. Поради необичайно развитие и преждевременно сливане (осификация) на костите се наблюдават и други аномалии в костите. В допълнение се наблюдава и деформацията на Маделунг, а някои от пациентите могат да получат прогресивна дегенерация, скованост, и болка в лакътните стави (остеоартрит).
По време на ранното детство, индивидите с акромезомелична дисплазия може също да покажат симптоми на аномалии на прешлените и да получат патологично изкривяване на гръбначния стълб.
В редки случаи може да има и други допълнителни аномалии. Пример за това са забавен пубертет и заболявания на роговицата.
Смята се, че има пет вида акромезомелична дисплазия в зависимост от мутацията в гените. Всеки от тях е изключително рядък и се наследява по автозомно-рецесивен механизъм, с изключение на типа Osebold-Remondini, който се унаследява автозомно-доминантно.
Видовете акромезомелична дисплазия са:
- Осеболд-Ремодини (Osebold-Remondini)
- Маротео (Maroteaux)
- Гребе (Grebe)
- Хънтър-Томпсън (Hunter-Thompson)
- Дю-Пан (Du Pan)
Типът Маротео се дължи на аномалия на 9-та хромозома (9p13-12). Гребе, Хънтър-Томпсън и Дю Пан се дължат на аномалия в 20-та хромозома (20q11.2). За съжаление все още не е открита къде точно е аномалията при Осеболд-Ремодини.
До 2005 г. в медицинската литература са описани около 10 случая на засегнати лица от тип Хънтър-Томпсън и около 40 до 50 лица, страдащи от тип Маротео. Броят на индивидите с тип Grebe не е известен, но се смята, че този тип на акромезомеличната дисплазия е почти изцяло представен от лица, живеещи в Бразилия.
Диагностиката на акромезомеличната дисплазия се основава на внимателна анамнеза, физикален преглед и образно-диагностични изследвания. Въпреки че ръцете и краката могат да изглеждат необичайно къси и широки при раждането, прогресивните аномалии, свързани с разстройството (необичайното скъсяване на костите на предмишниците и подбедриците, ниския ръст, аномалиите на прешлените, лакътните стави и др.) обикновено не се изявяват симптоматично до ранното детство.
Специализираните рентгенологични изследвания могат да потвърдят необичайното развитие и преждевременното сливане на диафизите. Освен това те могат да разкрият необичайно сливане на нарастващите краища на костите в пръстите, ръцете и краката. Такива изследвания могат също така да потвърдят наличието и/или степента на получените костни аномалии, както и други скелетни аномалии, които могат да бъдат свързани с акромезомелична дисплазия.
 Лечението на това заболяване е насочено към специфичните симптоми и физическите особености, видими при всеки индивид. Анормалната кривина на гръбначния стълб може да бъде лекувана с комбинация от упражнения и физиотерапия, скоби, корсети или в много тежки случаи с операция. Ранната намеса е важна, за да се гарантира, че децата със заболяването ще достигат своя потенциал на растеж.
Лечението на това заболяване е насочено към специфичните симптоми и физическите особености, видими при всеки индивид. Анормалната кривина на гръбначния стълб може да бъде лекувана с комбинация от упражнения и физиотерапия, скоби, корсети или в много тежки случаи с операция. Ранната намеса е важна, за да се гарантира, че децата със заболяването ще достигат своя потенциал на растеж.
Генетичното консултиране се препоръчва за засегнатите лица и техните семейства.
- Дисплазия на Астли-Кендал (Astley-Kendall Syndrome)
 Дисплазията на Астли-Кендал също е част от тази група заболявания. Този вид дисплазия е рядко заболяване, което засяга костите на човека. Синдромът на Астли-Кендал причинява увреждания в костите на човека, в резултат на което те стават много крехки и лесно чупливи.
Дисплазията на Астли-Кендал също е част от тази група заболявания. Този вид дисплазия е рядко заболяване, което засяга костите на човека. Синдромът на Астли-Кендал причинява увреждания в костите на човека, в резултат на което те стават много крехки и лесно чупливи.
Засегнатите лица също имат скъсени крайници и аномалии на хрущялната тъкан. В някои случаи синдромът Астли-Кендал може да доведе до мъртво раждане или непосредствена смърт след раждането.
Диагностиката на този вид дисплазия се поставя след преглед и снемане на анамнеза от страна на лекаря, както и образно-диагностични изследвания.
Лечение за това заболяване няма. Терапията е насочена към симптомите на заболяването, а именно нарушенията в здравината на костите. Препоръчват се медикаменти, които подобряват костната структура.
- Кампомеличен синдром
 Кампомеличният синдром е рядко вродено заболяване, при което има множество аномалии, засягащи главно костно-ставната система. Характеризира се с:
Кампомеличният синдром е рядко вродено заболяване, при което има множество аномалии, засягащи главно костно-ставната система. Характеризира се с:
- изкривяване на дългите кости на краката
- множество аномалии на лицето и небцето
- аномалии на рамото и таза
- единадесет чифта ребра вместо обичайните дванадесет
- неправилно развитие на трахеята
- забавяне на развитието или пълното отсъствие на гениталиите при мъжете
Кампомеличният синдром е рядка форма на скелетна дисплазия, характеризираща се с изкривяване на дългите кости на краката. Тазът и рамото могат да бъдат недоразвити. Черепът може да бъде патологично уголемен, издължен и тесен. Лицето може да изглежда плоско, с голямо чело, малка брадичка и цепнатина на небцето. Бебетата често изпитват респираторни проблеми и страдат от заболявания на дихателните пътища.
Респираторните проблеми, дължащи се на недоразвит гръден кош, са най-сериозните симптом на този синдром. Белите дробове може да не разполагат с достатъчно пространство, за да растат правилно поради аномалии в гръбначния стълб.
Други симптоми, които могат да се проявят при някои пациенти с кампомеличен синдром, са дислокация (разместване) на бедрата, недоразвити бели дробове, анормални прешлени и аномалии на сърцето и бъбреците. Някои индивиди, страдащи от синдрома, имат полови аномалии. Наблюдават се мъже с женски гениталии и женска репродуктивна система.
Смята се, че кампомеличният синдром се унаследява по автозомно-доминантен модел. Съществуват сведения за семейства, в които са засегнати голям брой деца, без да има видимо засягане на родителите. Това може да се дължи на единия родител, който има както нормални, така и анормални SOX9 гени, които са причината за аномалиите. В резултат на това едно или повече от децата на този родител могат да наследят генната мутация и да проявят нарушението, въпреки че родителят няма видими симптоми.
 Кампомеличният синдром е рядко заболяване, за което се смята, че засяга жените два пъти по-често от мъжете. Приблизително 100 случая на това разстройство са описани в медицинската литература.
Кампомеличният синдром е рядко заболяване, за което се смята, че засяга жените два пъти по-често от мъжете. Приблизително 100 случая на това разстройство са описани в медицинската литература.
Диагностиката се основава на клиничен преглед, рентегологично изследване на прешлени, таз, гръден кош и крака, както и ехографско изследване на бъбреците и ехокардиография на сърцето. ДНК анализът на кръвта също може да потвърди мутация в гена SOX9.
Лечение за заболяването няма. Терапията при кампомеличният синдром е по-скоро насочена към характерните симптоми и придружаващите заболявания. Тя се състои от механична или физическа помощ при дишане, както и ортопедични медицински грижи и операции.
- Хондродисплазия с охлювоподобен таз (chondrodysplasia with snail-like pelvis)
 Хондродисплазия с охлювоподобен таз или т.нар. Schneckenbecken dysplasia представлява много рядко генетично разстройство, включващо анормално развитие на костите и хрущялите, което се изразява с:
Хондродисплазия с охлювоподобен таз или т.нар. Schneckenbecken dysplasia представлява много рядко генетично разстройство, включващо анормално развитие на костите и хрущялите, което се изразява с:
- разцепване на небцето
- скъсен врат
- нисък ръст
- необичайна охлювоподобна форма на таза
Състоянието обикновено води до мъртво раждане или смърт скоро след раждането.
Характерните симптоми и особености на заболяването са:
- Излишък на амниотична течност (биологичен продукт, съдържащ се в околоплодния мехур и помага с развитието на бебето)
- Оток
- Голяма глава
- Плоско лице
- Разцепване на небцето
- Къс врат
- Нарушения в костната структура
- Таз с формата на охлюв
- Къси ребра
- Нисък ръст
Причината за тази аномалия не е ясна. Предполага се, че се дължи на мутация в някой от гените, но все още не е уточнено кой точно.
Диагностиката на този вид хондродисплазия се извършва чрез физикален преглед и образно-диагностични изследвания като рентгенография и компютърна-томография.
Лечение за хондродисплазията с охлювоподобен таз няма, а прогнозата е неблагоприятна.
- Синдром на Уолкот-Ралисон
Синдромът на Уолкот-Ралисон е последното рядко заболяване от групата на друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб. Характеризира се с неонатален диабет, множествена дисплазия на костите, както и повтарящи се епизоди на остра чернодробна недостатъчност.
 По-малко от 60 случая са регистрирани досега, а разпространението може да варира значително между отделните страни.
По-малко от 60 случая са регистрирани досега, а разпространението може да варира значително между отделните страни.
Както вече беше споменато основните симптоми на синдромът на Уолкот-Ралисон са ранният диабет, скелетната дисплазия и уврежданията в чернодробната функция.
Диабетът се развива рано, обикновено преди шест месечна възраст, и е диабет тип 1.
Скелетната дисплазия обикновено се проявява в първата или втората година от живота и се свързва с нисък ръст (джудже). Промените, засягащи дългите кости, таза и прешлените могат да се видят на рентгенологичен образ още от началото на диабета.
Чернодробната дисфункция е най-животозастрашаващото усложнение и се проявява от повишени чернодробни ензими, уголемяване на черния дроб и повтаряща се остра чернодробна недостатъчност.
Други прояви варират между пациентите и включват бъбречна дисфункция, панкреасна недостатъчност, интелектуален дефицит, хипотиреоидизъм, и постоянни инфекции.
Синдромът се причинява от мутации в гена EIF2AK3, кодиращ еукариотния фактор за иницииране на транслация 2-алфа киназа 3, която играе ключова роля в контрола на транслацията по време на синтеза на протеини в тялото.
транслацията по време на синтеза на протеини в тялото.
Диагнозата се поставя след физикален преглед и представяне на симптомите. Винаги трябва да се мисли за синдром на Уолкот-Ралисон при всяко кърмаче с неонатален диабет и скелетна дисплазия и/или епизоди на остра чернодробна недостатъчност. На рентгенография се виждат ранни признаци на множествена дисплазия и дефицитна минерализация на костите. Молекулярно-генетичното изследване потвърждава диагнозата.
Лечението на синдрома на Уолкот-Ралисон е насочено главно към ранния диабет и чернодробната недостатъчност, както и лечението на други придружаващи заболявания или синдроми.
Прогнозата е лоша за повечето пациенти, защото се наблюдава голям процент смъртност, причинена от органна недостатъчност.
Симптоми и признаци при Друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб МКБ Q77.8
- Ментална ретардация
- Затруднено дишане
- Нисък ръст
- Симптоми на сърцето
- Мускулна слабост
- Симптоми на челюстта
Библиография
https://rarediseases.org/rare-diseases/metatropic-dysplasia-i/
https://ghr.nlm.nih.gov/condition/metatropic-dysplasia
https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=2635
https://rarediseases.org/rare-diseases/leri-weilldyschondrosteosis/
https://rarediseases.info.nih.gov/diseases/3224/leri-weill-dyschondrosteosis
https://ghr.nlm.nih.gov/condition/leri-weill-dyschondrosteosis
https://rarediseases.info.nih.gov/diseases/6/acromesomelic-dysplasia
https://rarediseases.org/rare-diseases/acromesomelic-dysplasia/
https://rarediseases.info.nih.gov/diseases/9220/astley-kendall-syndrome
http://www.diseaseinfosearch.org/Astley-Kendall+Syndrome/634
http://www.checkorphan.org/diseases/astley-kendall-syndrome
https://ghr.nlm.nih.gov/condition/campomelic-dysplasia
https://rarediseases.info.nih.gov/diseases/10027/campomelic-dysplasia
https://rarediseases.org/rare-diseases/campomelic-syndrome/
https://rarediseases.info.nih.gov/diseases/169/schneckenbecken-dysplasia
https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=3144
http://www.checkorphan.org/diseases/schneckenbecken-dysplasia
https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=1667
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2991281
Коментари към Друга остеохондродисплазия с дефекти в растежа на тръбестите кости и гръбначния стълб МКБ Q77.8