Хондроектодермална дисплазия МКБ Q77.6
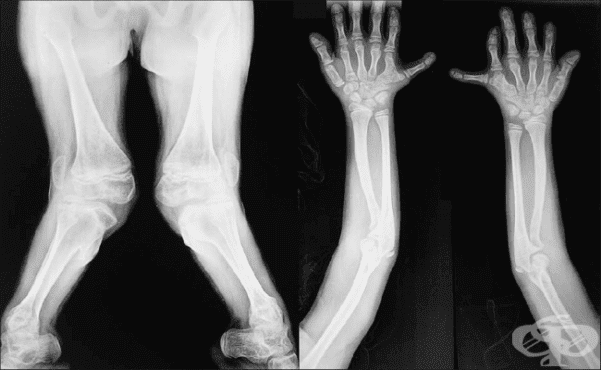
Хондроектодермалната дисплазия, наречена още синдром на Елис ван Кревелд (Ellis van Creveld Syndrome), представлява рядко наследствено заболяване, което води до много нисък ръст. Хората с тази аномалия имат много къси предмишници, подбедрици, къси ребра с тесен гръден кош.
 Синдромът на Ellis-van Creveld е описан за пръв път от лекарите Ричард У. Б. Елис и Симон ван Кревелд. В края на 30-те години двамата педиатри се срещат във влак, докато пътуват до конференция по педиатрия в Англия и откриват, че всеки от тях има пациент с подобни изяви. През 1940 г. те официално описват генетичния синдром, който носи техните имена, въпреки че първоначално го нарекли хондроектодермална дисплазия или мезоектодермална дисплазия.
Синдромът на Ellis-van Creveld е описан за пръв път от лекарите Ричард У. Б. Елис и Симон ван Кревелд. В края на 30-те години двамата педиатри се срещат във влак, докато пътуват до конференция по педиатрия в Англия и откриват, че всеки от тях има пациент с подобни изяви. През 1940 г. те официално описват генетичния синдром, който носи техните имена, въпреки че първоначално го нарекли хондроектодермална дисплазия или мезоектодермална дисплазия.
Честотата над хондроектодермалната дисплазия е 1:200 000.
Според учените синдромът на Ellis-Van Creveld се дължи на мутация в два гена, наречени EVC и EVC2. Тези генни мутации водят до производството на видоизменени EVC и EVC2 протеини. Точната функция на протеините все още не е изяснена, но се предполага, че е важна за изграждането на костите и зъбите, както и за нормалния растеж и развитие. Някои засегнати индивиди нямат мутации в тези гени, така че е вероятно и други гени да носят отговорност за EVC протеините.
Хондроектодермалната дисплазия се наследява по автозомно-рецесивен модел. Рецесивните генетични нарушения се появяват, когато човек наследи две копия на променен ген, по едно копие от всеки родител. Ако дадено лице получи един нормален и един променен ген, лицето ще бъде само здрав носител на болестта. Родителите на индивид с автономно-рецесивно заболяване носят по едно копие на мутиралия ген, но те нямат признаци и симптоми на болестта, тоест са здрави носители.
При индивидите с хондроектодермалната дисплазия обикновено се наблюдават необичайно скъсени ръце и крака, докато размерите на главата и торса са нормални. Допълнителни пръсти (полидактилия) присъстват при всички пациенти с това заболяване и обикновено са засегнати и двете ръце. Аномалиите включват също и анормално развитие на косата, ноктите и зъбите.
Повече от 50% от пациентите със синдром на Елис ван Кревелд се раждат с малформации на сърцето. Най-честият сърдечен дефект е наличие на необичайно отваряне в стената между двете предсърдия (междупредсърден септален дефект). Съобщени са и други видове сърдечни дефекти, включително дефекти на междукамерната преграда и Боталов проток.
При някои момчета, страдащи от вродената аномалия, се наблюдава и крипторхизъм или нарушения в пениса. Наблюдавани са и аномалии на гръдната стена, гръбначния стълб и дихателната система.
Клинични симптоми при вродената аномалия включват:
- Разцепване на устната и небцето (заешка устна, вълча уста)
- Еписпадия
- Крипторхизъм
- Полидактилия
- Проблеми с ноктите, включително деформирани и липсващи нокти
- Къси ръце и крака
- Нисък ръст 1-1,5 метра
- Рядка на места липсваща коса
- Аномалии на зъбите - широко разположени, липсващи зъби и др.
- Сърдечни малформации
Пренатални аномалии могат да бъдат открити още през 18-та гестационна седмица чрез ултразвуково изследване.
На рентгенологичен образ при хондроектодермалната дисплазия се наблюдава прогресивно дистално скъсяване на дългите кости. В ранна детска възраст често срещана е дисплазията на тазобедрената става.
Диагнозата синдром на Елис ван Кревелд може да бъде поставена чрез:
- Ултразвуково изследване
- Рентгенография на гръден кош
- Ехокардиография
- Генетично изследване за установяване на мутации в един от двата гена (EVC, EVC2)
 Генетичното заболяване може да се открие пренатално чрез ултразвуково изследване. Откриването на няколко структурни дефекта на плода в края на първия триместър — тесен гръден кош, къси и извити дълги кости, сърдечен дефект, може да насочва към диагнозата синдром на Ellis van Creveld. В този период е важно синдрома да се отграничи от синдрома на късото ребро (Синдром на Жун) и синдромите на полидактилия с къси ребра.
Генетичното заболяване може да се открие пренатално чрез ултразвуково изследване. Откриването на няколко структурни дефекта на плода в края на първия триместър — тесен гръден кош, къси и извити дълги кости, сърдечен дефект, може да насочва към диагнозата синдром на Ellis van Creveld. В този период е важно синдрома да се отграничи от синдрома на късото ребро (Синдром на Жун) и синдромите на полидактилия с къси ребра.
Лечението при вродената аномалия зависи от това коя система е засегната и от неговата тежест. Генетичното консултиране се препоръчва на засегнатите деца и техните семейства.
Лечението за синдрома е предимно насочено срещу симптомите на аномалията, включващо лечение на дихателната недостатъчност, дължаща се на тесния гръден кош и лечение на сърдечна недостатъчност, поради вродените сърдечни дефекти.
Ортопедичните корекции при този синдром имат за цел коригиране на полидактилията или други малформации. Костните деформации и нарушенията в ставите, както и опорно-двигателния апарат, изискват непрекъснато ортопедично проследяване и грижи.
Може да е необходима и сърдечна операция за коригиране на сърдечните аномалии. Лечението на вродени сърдечни малформации при пациенти с  хондроектодермална дисплазия е свързано със значителен риск от смърт.
хондроектодермална дисплазия е свързано със значителен риск от смърт.
В литературата при тази аномалия се описват и операции за отстраняване на урологичните и дихателните аномалии.
Продължителността на живота е намалена главно поради тежестта на дихателната недостатъчност, както и вроденото сърдечно заболяване.
Прогнозата при синдрома на Ellis van Creveld е неблагоприятна. Близо 50% от бебетата умират в ранна детска възраст, вследствие на сърдечно-респираторни усложнения.
Симптоми и признаци при Хондроектодермална дисплазия МКБ Q77.6
- Нисък ръст
- Симптоми от пръстите
- Стоматологични симптоми
- Симптоми от пръстите на краката
- Липсващи зъби
- Симптоми, свързани с промяна на ноктите
Лечение на Хондроектодермална дисплазия МКБ Q77.6
Библиография
https://ghr.nlm.nih.gov/condition/ellis-van-creveld-syndrome
https://en.wikipedia.org/wiki/Ellis%E2%80%93van_Creveld_syndrome
https://rarediseases.org/rare-diseases/ellis-van-creveld-syndrome/
https://radiopaedia.org/articles/chondroectodermal-dysplasia
Коментари към Хондроектодермална дисплазия МКБ Q77.6