Болест на Hallervorden-Spatz МКБ G23.0
› История
› Диагноза
› Лечение
› Прогноза
Пантотенат киназа-асоциирана невродегенерация, ПКАН (Pantothenate kinase-associated neurodegeneration, PKAN), наричана преди болест на Hallervorden-Spatz, е рядко генетично заболяване, характеризиращо се с прогресивна екстрапирамидна дисфункция и деменция.
Заболяването е описано за първи път през 1922 г. от двама немски лекари, Hallervorden и Spatz, като форма на фамилна мозъчна дегенерация, характеризираща се с церебрално отлагане на желязо. Терминът невродегенерация с натрупване на желязо в мозъка тип 1 (NBIA-1) в крайна сметка започва да се използва за това състояние, въпреки че най-новият термин за разстройството е пантотенат киназа-асоциирана невродегенерация.
Началото на симптомите най-често се появява в края на детството или ранното юношество. Класическото представяне е в края на първото десетилетие или в началото на второто десетилетие, на възраст между 7 и 15 години. Въпреки това, заболяването се съобщава в ранна детска възраст, а също така са описани случаи с начало в зряла възраст.
ПКАН е подтип на невродегенерация с натрупване на желязо в мозъка (NBIA). NBIA включва група от наследствени невродегенеративни заболявания, които се характеризират симптоматично с екстрапирамидни двигателни нарушения и патологично с анормално натрупване на желязо в дълбоки базални ганглии.
Досега са описани няколко форми и всяка има съответен ген и специфичен начин на наследяване:
- ПКАН, асоциирана с PLA2G6 невродегенерация (PLAN)
- невродегенерации, свързани с хидроксилаза на мастни киселини (FAHN)
- невродегенерации, свързани с коензим А протеин синтаза (CoPAN)
- невродегенерации, свързани с протеини на митохондриалната мембрана (MPAN)
- синдром на Kufor-Rakeb
- синдромът на Woodhouse-Sakati
- ацерулоплазминемия
Заболяването е с изразена прогресивност.
История
Заболяването е описано за първи път от Hallervorden и Spatz през 1922 година. Тяхното откритие е предизвикано от диагноза на семейство, в което пет сестри показват прогресивно нарастваща деменция и дизартрия.
Аутопсиите разкриват кафяви промени в цвета в различни области на мозъка (особен интерес представляват областите globus pallidus и substantia nigra).
По-нататъшно изследване и описание е направено от Meyer през 1958 година, който диагностицира 30 отделни случая на заболяването. През 1978 Elejalde описва засегнати членове на семейство и изказва хипотезата, че състоянието произхожда от Централна Европа, подкрепяйки хипотезата си с клиничен и генетичен анализ.
Допълнителни изследвания са предоставени от Malmstrom-Groth и Kristensson през 1982 година и Jankovic през 1985 година.
Генът на заболяването е локализиран в хромозома 20p от Taylor през 1996 г., който предлага това състояние да се нарича невродегенерация с натрупване на желязо в мозъка (NBIA1), за да се избегне епонима на Hallervorden-Spatz. Заболяването е преименувано на пантотенат киназа-асоциирана невродегенерация (ПКАН) от Zhou през 2001 г., който предлага термина, за да избегне погрешно тълкуване и да отрази по-добре истинската природа на заболяването.
Епидемиология
Данните за разпространението по отношение на това заболяване остават непълни, но се изчислява, че някъде между 1 на 1 000 000 до 3 на 1 000 000 индивида ще бъдат засегнати от това състояние (въз основа на наблюдавани случаи в популация), но отново това е само оценка, тъй като заболяването е толкова рядко, че е трудно да се установи точно статистика.
Етиология
Точната етиология на ПКАН не е известна. Една предложена хипотеза е, че анормалната пероксидация на липофусцин до невромеланин и дефицитната цистеин диоксигеназа водят до анормално натрупване на желязо в мозъка. Докато части от globus pallidus и pars reticulata на substantia nigra имат високо съдържание на желязо при здрави индивиди, индивидите с ПКАН имат излишни количества желязо, отложени в тези области.
Въпреки това, точната роля на желязото в етиологията на това заболяване остава неизвестна. Освен това не е ясно дали отлагането на желязо в базалните ганглии при ПКАН е причина или следствие от загуба на неврони и глиоза. Намалена активност на ензима цистеин диоксигеназа е демонстрирана при едно засегнато дете. Предполага се, че това води до натрупване на цистеин в базалните ганглии, тъй като цистеинът може да хелатира желязото и по този начин да доведе до неговото отлагане. Тези констатации обаче не са потвърдени при възрастни пациенти.
Предположена е роля на мутацията в гена PANK2 (лента 20p13) в етиологията на заболяването. Дефицитът на пантотенат киназа може да доведе до натрупване на цистеин и цистеин-съдържащи съединения в базалните ганглии. Това причинява хелатиране на желязото в глобус палидус и генериране на свободни радикали в резултат на бързо автоокисление на цистеин в присъствието на желязо. Мутациите в гена PANK2 са причина за повечето наследени случаи на болест на Hallervorden-Spatz. Такива мутации водят до автозомно-рецесивна вродена грешка на метаболизма на коензим А, която е наречена невродегенерация, свързана с пантотенат киназа.
Патоморфология
Патологичното изследване разкрива характерно ръждиво-кафяво оцветяване на globus pallidus и вторично на отлагането на желязо. Може да се отбележи генерализирана атрофия на мозъка с намаляване на размера на nucleus caudatus, substantia nigra и тегментума. Микроскопски характерните промени включват:
- променлива загуба на неврони, миелинизирани влакна и глиоза в globus pallidus и substantia nigra, които могат да изглеждат спонгиозни, когато са тежки
- широко разпространени кръгли или овални, безядрени структури, наречени сфероиди (известни също като невроаксонална дистрофия). Представляват аксони с вакуолизирана цитоплазма и се намират най-често в палидонигралната система, въпреки че присъстват и в мозъчната кора
- натрупване на пигмент, съдържащ предимно желязо, в globus pallidus и substantia nigra
- липофусцин и невромеланин, съдържащи желязо, в основно засегнатите области.
Отлагането на желязо може да се открие вътреклетъчно и извънклетъчно и често е концентрирано в съдовете. Тези промени се откриват в по-малка степен в други части на мозъка и в гръбначния мозък. Наличието на аксонални сфероиди предполага връзка между ПКАН и инфантилна невроаксонална дистрофия, въпреки че не се съобщава за клинична или генетична връзка между двете заболявания. Тау-положителни неврофибриларни възли и алфа-синуклеин-позитивни телца на Lewy могат да бъдат открити в кортикалните и субкортикалните региони при пациенти с продължително клинично протичане.
Генетични характеристики
Пантотенат киназа-асоциирана невродегенерация (ПКАН) е автозомно-рецесивно заболяване. И двамата родители на засегнато дете трябва да са хетерозиготни носители на болестта и следователно трябва да носят един мутирал алел. Тъй като това е автозомно разстройство, тези хетерозиготни за заболяването индивиди може да не показват никакви атипични характеристики, които се считат за предполагащи разстройството, но има докладвани случаи на комбинирана хетерозиготност, при които хетерозиготни индивиди развиват класическата форма на заболяването.
Състоянието се причинява от мутантен PANK2 ген, разположен в хромозомния локус: 20p13-p12.3. PANK2 е отговорен за кодирането на протеина пантотенат киназа 2. PANK2 кодира ензима пантотенат киназа и мутациите в гена водят до вродена грешка в метаболизма на витамин В5 (пантотенат). Витамин В5 е необходим за производството на коензим А в клетките. Разрушаването на този ензим засяга енергийния и липидния метаболизъм и може да доведе до натрупване на потенциално вредни съединения в мозъка, включително желязо.
Генът PANK2 също кодира 50,5-kDaпротеин, който е функционална пантотенат киназа, важен регулаторен ензим в биосинтезата на коензим А (CoA) и катализиращ фосфорилирането на пантотенат (витамин B5), N-пантотеноил-цистеин и пантетеин.
Това заболяване е докладвано в специфични общности, базирани на бракове в рамките на общността, където и двамата родители на детето са носители на една и съща мутация.
Клинична картина
Симптомите при пантотенат киназа-асоциирана невродегенерация (ПКАН) включват:
- дистония - видна и ранна характеристика
- значителни нарушения на говора - могат да се появят рано
- дисфагия - често срещан симптом, причинени от ригидност и кортикобулбарно засягане
- деменция - присъства при повечето индивиди с ПКАН
- зрително увреждане - причинено от оптична атрофия или дегенерация на ретината. Не е необичайно и може да бъде симптом на заболяването, въпреки че е рядко
- припадъци - не често
Клиничните прояви варират при отделните пациенти. Симптомите обикновено започват през първото десетилетие с двигателно разстройство от екстрапирамиден тип и затруднена походка. Екстрапирамидните симптоми доминират в клиничната картина и включват ригидност, забавени движения, дистония, хореоатетоза и тремор.
При пациенти с атипичен ПКАН, екстрапирамидната дисфункция може да се забави с няколко години и спастичността и дизартрията могат да бъдат симптомите.
Физикалният преглед разкрива признаци, съответстващи на екстрапирамидна и кортикоспинална дисфункция. В допълнение към ригидност, дистония и хорея, пациентите могат да проявят спастичност, оживени рефлекси и екстензорни плантарни реакции.
Въз основа на общите клинични характеристики са предложени следните диагностични критерии за ПКАН. За окончателна диагноза трябва да са налице всички задължителни находки и поне 2 от потвърждаващите находки. Не трябва да присъства нито един от изключващите фактори.
Задължителните характеристики на ПКАН включват:
- начало през първите 2 десетилетия от живота
- прогресия на признаците и симптомите
- доказателства за екстрапирамидна дисфункция, включително 1 или повече от следните: дистония, ригидност, хореоатетоза
Потвърждаващите характеристики включват:
- засягане на кортикоспиналния тракт
- прогресивно интелектуално увреждане
- пигментен ретинит и/или оптична атрофия
- гърчове
- положителна фамилна анамнеза, съответстваща на автозомно рецесивно унаследяване
- хипоинтензни зони на ядрено-магнитен резонанс (ЯМР), включващи базалните ганглии
- анормални цитозоми в циркулиращите лимфоцити и/или морскосини хистиоцити в костния мозък
Изключващите характеристики включват
- абнормени нива на церулоплазмин и/или аномалии в метаболизма на медта
- наличие на явна невронална цероидна липофусциноза, демонстрирана от тежко зрително увреждане и/или гърчове, които са трудни за контролиране
- преобладаващите епилептични симптоми
- тежка дегенерация на ретината или зрително увреждане, предхождащи други симптоми
- наличие на фамилна анамнеза за хорея на Хънтингтън и/или други автозомни, доминантно наследствени невродвигателни разстройства
- наличие на атрофия на каудатус при образни изследвания
- дефицит на хексозаминидаза А
- дефицит на ганглиозид моносиалова киселина-1 (GM1)–галактозидаза
- липса на екстрапирамидни признаци
Пациентите с атипичен ПКАН показват по-разнообразни клинични характеристики с по-бавна прогресия. Представящите се симптоми обикновено включват говорни дефекти, като палилалия (повтаряне на думи или фрази), тахилалия/тахилогия (бърза реч) и дизартрия (лоша артикулация, неразбираема реч). Началото е през първите три десетилетия (средна възраст 13,6 години).
Около 25% от хората развиват нехарактерна форма на ПКАН, която се развива след 10-годишна възраст и следва по-бавно, по-постепенно темпо на влошаване в сравнение с тези на възраст преди 10 години. Тези индивиди са изправени пред значителни говорни дефицити, както и психиатрични и поведенчески смущения.
Като прогресивно, дегенеративно нервно заболяване, болестта на Hallervorden-Spatz води до ранна неподвижност и често смърт в ранна зряла възраст. Смъртта настъпва преждевременно поради инфекции като пневмония и заболяването само по себе си не ограничава живота.
Диагноза
Образна диагностика
Компютърна томография
Компютърната томография (КТ) не е много полезна при диагностицирането на ПКАН, но може да покаже хиподенситет в базалните ганглии и известна атрофия на мозъка. Описано е и калцифициране в базалните ганглии при липса на каквато и да е атрофия.
Еднофотонна емисионна компютърна томография (SPECT)
Йод-123 (123 I)-бета-карбометокси-3бета-(4-флуорофенил) тропан (CIT) сканиране с еднофотонна емисия компютърна томография (SPECT) и (123 I)-йодобензамид (IBZM)-SPECT сканиране също са били използвани при поставяне на диагнозата ПКАН.
Ядрено-магнитен резонанс (ЯМР)
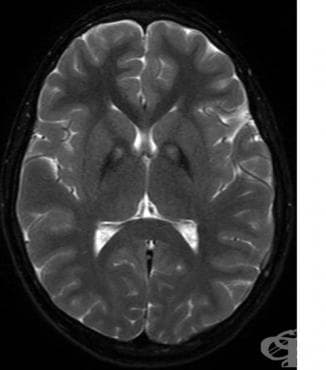
ЯМР е увеличил вероятността за предсмъртно диагностициране на ПКАН. Установяват се двустранно симетрични, хиперинтензивни промени на сигнала в предния медиален глобус палидус, със заобикаляща хипоинтензивност в глобус палидус. Тези образни характеристики са доста диагностични за ПКАН и са наречени „окото на тигъра“.
Установено е, пациентите с ПКАН имат повече от 3 пъти по-висока концентрация на желязо в глобус палидус, субталамично ядро и вътрешна капсула, в сравнение с нормалните контроли. Пациентите, хетерозиготни за PANK2 мутацията, не са имали никакви находки за отлагане на желязо.
Лабораторни изследвания
Молекулярното генетично изследване включва тестване за PANK2 чрез анализ на последователност или делеция/дупликация. Когато един патогенен вариант (вариант на последователност или делеция на частичен или цял ген) се идентифицира при човек със знак "око на тигъра" на ЯМР, диагнозата болест на Hallervorden-Spatz се потвърждава.
Невропатология
Микроскопските характеристики на пантотенат киназа-асоциирана невродегенерация (ПКАН) включват високи нива на желязо в globus pallidus и pars reticulata на substantia nigra, липофусцин и невромеланин, концентриран в областите, натрупващи желязо, овални, безядрени структури, представляващи увеличени аксони, чиято цитоплазма набъбва с вакуоли, наричани сфероиди, невроаксонална дистрофия и тела на Леви.
Лечение
Исторически погледнато, лечението на пациенти с пантотенат киназа-асоциирана невродегенерация (ПКАН), е било предимно симптоматично. Но нови терапии са изследвани като потенциални агенти, модифициращи заболяването. Те включват хелатори на желязо, както и фосметпантотенат.
По отношение на симптоматичната терапия треморът се повлиява най-добре от допаминергични средства. Антихолинергичният агент бензтропин може да се използва за подпомагане на ригидност и тремор. Бензодиазепините са били изпробвани за хореоатетотични движения.
Хипертонията обикновено е комбинация от ригидност и спастичност и може да бъде трудна за лечение. Допаминовите агонисти и антихолинергиците могат да помогнат за намаляване на ригидността.
Баклофенът в умерени дози облекчава сковаността и спазмите и може да намали дистонията. Интрамускулен ботулинов токсин също е използван при възрастни, както и при деца.
Дълбока мозъчна стимулация, по-специално насочена към двустранните субталамични ядра, е изпробвана при пациенти с изразени апендикуларни симптоми. Симптоми като хиперсаливация и дизартрия могат да бъдат проблемни, като лекарства като метскополамин бромид могат да бъдат изпробвани за прекомерно слюноотделяне.
Дизартрията може да реагира на лекарства, използвани за ригидност и спастичност. Логопедичната терапия също може да бъде полезна, а в напреднали случаи могат да се използват компютърно подпомагани устройства.
Може да се наложи хранене чрез гастростома, когато дисфагията прогресира.
Системни хелатиращи агенти, като десфериоксамин, са били използвани в опит да се премахне излишното желязо от мозъка, но те не са се оказали полезни. Деменцията е прогресираща и нито едно лечение не е доказано ефективно.
In vitro, плурипотентни стволови клетки, получени от пациенти с ПКАН, реагират на прилагането на коензим А чрез предотвратяване на загиването на невроните и образуването на реактивни кислородни радикали.
Лечение на дистония
Дистонията е най-изявеният и инвалидизиращ симптом на ПКАН и може да реагира умерено на допаминергични агенти като леводопа и бромокриптин (допаминов агонист). Други допаминови агонисти, като ропинирол или прамипексол, също могат да бъдат обмислени, въпреки че не са провеждани официални проучвания за тяхната ефикасност при това заболяване.
Антихолинергичните средства, като трихексифенидил, могат да се използват, когато допаминергичните средства не са полезни. Въпреки това, тези лекарства носят само преходно облекчение за дистония, а физиотерапията също често е от ограничена полза. Ботулиновият токсин може да се инжектира в силно засегнати мускули за облекчаване на дистонията.
Хирургично лечение
Тъй като дистонията е важна характеристика на ПКАН, глобус палидус е цел за хирургично лечение. Стереотактична палидотомия и двустранна таламотомия понякога се опитват при пациенти с тежка дистония, което води до частично облекчаване на симптомите. Дълбока мозъчна стимулация на globus pallidus, както и субталамичното ядро е използвана при тези пациенти с обещаващи резултати.
Продължителна интратекална инфузия на баклофен е изпробвана за рефрактерна генерализирана дистония без особен успех. Алтернатива е интравентрикуларен баклофен, който представлява интерес, тъй като това място може да донесе полза при пациенти, които имат основно дистония на горната част на тялото и лицето, като блефароспазъм.
Диетичен режим
Диетата може да играе роля при лечението на болест на Hallervorden-Spatz. Изследване, проведено върху група братя с ПКАН, показва положителен отговор на хиперкалорична диета. Братята са били поставени на диета от 50 kcal/kg в продължение на 2 седмици и е отбелязано, че и двамата имат подобрение, особено по отношение на дистонията на шията и торса, както и с походката и силата на захвата на ръцете.
Прогноза
Клиничното протичане на ПКАН е променливо в зависимост от неговата форма (класическа или атипична). При пациенти с класическа форма заболяването има прогресивен ход, продължаващ няколко години, водещ до смърт в ранна детска възраст.
Някои пациенти изпитват бързо влошаване на функцията вследствие на дистония, ригидност, дисфагия и респираторни нарушения, като умират в рамките на 1-2 години от началото на заболяването.
Други пациенти претърпяват по-бавна прогресия или дори плато в продължение на много години и могат да продължат да функционират до третото десетилетие от живота.
Честотата на преживяемост за тези, диагностицирани с типичен ПКАН и оставени без лечение, е 11,18 години със стандартно отклонение от 7,8 години.
Коментари към Болест на Hallervorden-Spatz МКБ G23.0