Други вродени аномалии на костно-мускулната система МКБ Q79.8
Вродените аномалии на костно-мускулната система са рядко срещани. Появяват се вътреутробно и причините за тях са разнообразни — тератогенни фактори, някои лекарства, недостатъчен прием на фолиева киселина, инфекциозни причинители, генетични заболявания и нови мутации. Вродената липса на мускули е рядко заболяване с честота 1 на 11 000 живородени деца. Други вродени аномалии на костно-мускулната система включват:
- липса на мускул
- липса на сухожилие
- липса на кост
- добавъчен мускул
- вродена амиотрофия
- вродени стягащи ленти (амниотични ленти/амниотични върви)
- скъсяване на сухожилие
- синдром на Poland
Вродената липса на коремните мускули е рядка малформация. Липсата на цяла мускулна група е по-рядка от тази на отделните мускули, а двустранното засягане е по-рядко срещано от едностранното.
Най-често описаните дефекти са в гръдните мускули. Според някои автори описаните случай на липса на гръдни мускули са 42, според други — 112.
Вродена липса на глутеалните (седалищни) мускули е описано за първи път през 1976 г. Счита се, че е предизвикано от автозомно-рецесивен ген. Ако липсват глутеалните мускули, не биха били възможни ежедневни действия. Gluteus maximus отвежда бедрото в ханша при действия като изкачване на стълби, катерене или ходене, както и повдига задните части, и също така предотвратява движението на тялото напред или назад, когато останалата част от тялото се движи. Големият седалищен мускул също спомага за стабилизиране на бедрената кост и пищяла. Gluteus minimus et medius също са част от глутеалните мускули, но са по-малки. Ако тези мускули липсват, кракът няма да може да отведе или медиално да завърти бедрото. Тялото също няма да може да премества тежестта от едната страна на другата, когато единият крак е на земята.
Липсата на абдоминални мускули може да се дължи на хипертрофирал пикочен мехур. Издуването на мехура може да прекъсне въобще формирането на тези мускули или от повишеното вътрекоремно налягане, причинено от дилатацията му, може да доведе до атрофия на мускулите.
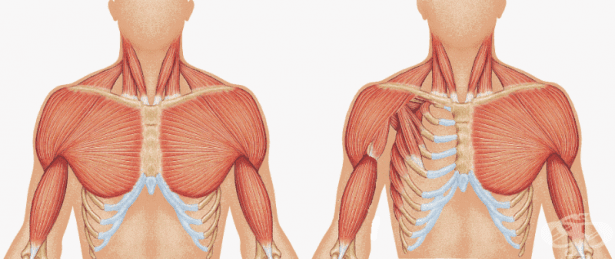
По-честа малформация на костно-мускулната система е липсата на гръдните мускули (pectoralis major et minor). Нарича се синдром на Поланд, наречен от британския хирург Алфред Поланд и представлява рядък зародишен дефект, характеризиращ се с недоразвитие или отсъствие на гръдния мускул (pectoralis) от едната страна на тялото, също и кожата на пръстите (кожна синдактилия) на ръката на същата страна. Честотата му е от 1:7000 до 1:100 000 души.
Причината за синдрома в Поланд е неизвестна. Подключичните артерии доставят кръв на ембрионалните тъкани, които изграждат гръдната стена и ръката. Различията в мястото и степента на нарушението могат да обяснят диапазона на признаците и симптомите, които се появяват в синдрома. Отклонения в ембрионалната структура, които помагат за ранното развитие на крайниците, също могат да бъдат включени в това разстройство.
При повечето засегнати индивиди липсващата част е голямата част от мускула, която обикновено се прикрепя към горната част на ръката от едната страна и гръдната кост от другата. Възможни са и други аномалии на засегнатата страна на гръдния кош. В някои случаи липсват или са слабо развити допълнителните мускули в гръдната стена и рамо. Тогава те са заместени от сухожилия.
Може да има и аномалии на гръдния кош, като по-къси ребра, които се забелязват по-лесно поради по-малкото количество мазнини под кожата (подкожна мастна тъкан). Възможно е също така да се появят аномалии на гърдите и зърната, а космите под мишниците (аксиларните) понякога са редки или необичайно разположени. В повечето случаи аномалиите в областта на гръдния кош не причиняват здравословни проблеми и не повлияват движението. Синдромът на Поланд най-често засяга дясната страна на тялото и се среща по-често при мъже, отколкото при жени. Обикновено се счита за едностранно условие. Придружаващите симптоми са брахидактилия (къси пръсти), разположено в дясната гръдна половина сърце, диафрагмална херния, дефект или липса на раменна кост, аномалии на чернодробните или жлъчни пътища, олигодактилия (липсващи пръсти), анормална или липсваща лъчева кост, липсваща или анормална ушна мида, асиметрия на горните крайници, анормални ребра.
Диагноза се поставя веднага след раждането, заради физическите характеристики, използват се и компютърна томография, рентгенография, ядрено-магнитен резонанс. Лечението включва поставянето на силиконов гръден имплант или липомоделиране-трансфер на мастна тъкан от една част към друга на тялото.
Липсата на подбедрени (прасец) мускули се наблюдава изключително рядко, като е описан един-единствен случай на липса на цял прасец.
Етиологията на липсата на коремна мускулатура е неизяснена. Случаите на вродено липса на прав коремен мускул (ректус мускул) са рядкост, а двустранните са още по-редки. Вродената липса на двустранни долни прави коремни мускули е още по-рядко и често се среща при черепно-фациална дисплазия.
Пациентите с вродена липса на коремна мускулатура имат аномалии на пикочния тракт и ако те са мъже, неспаднали тестиси. Аномалиите, засягащи сърдечно-съдовата, стомашно-чревната и мускулно-скелетната система, са чести, но променливи находки. Колагените отлагания, които заместват гладката мускулатура на уринарния тракт, дисплазията на бъбречния паренхим и недоразвитие на коремните мускули са основните хистологични характеристики. Неефективното изпразване на пикочните пътища предразполага тези пациенти да са по-податливи на уринарна инфекция, а нарушеният механизъм на кашлицата може да доведе до пневмония. Бъбречната недостатъчност и сепсисът са най-честите причини за смърт.
Към други вродените аномалии на костно-мускулната система се включват и липсващи кости. Причините отново са същите. Срещат се недоразвитие (хипоплазия) или пълна липса (аплазия) на кости на дланта, лъчевата и лакътна кост на предмишницата. Най-тежките дефекти са липсата на цели сегменти от крайника или на целия. Когато липсва предмишница или лакът, но е налице китка, се нарича фокомелия. Този тип ръка се нарича още тюленоподобен крайник. Възможно е да липсват кости на долния крайник, като една или повече тазови кости, поради липса на растеж (аплазия) или забавен растеж (хипоплазия).
Описани са случаи на пациенти с липсваща бедрена кост. Липсата е част от проксималния бедрен фокален дефицит (PFFD). Среща се изключително рядко с честота 0,2 на 10 000 живородени деца. PFFD включва широк спектър от малформации, вариращи от малка хипоплазия до пълна агенезия (липса) на бедрената кост.
Фибуларна хемимелия (надлъжен фибуларен дефицит) е вродено състояние на липса на фибулата (малък пищял) и е най-честата аномалия на липса на кост на долните крайници. Характеризира се с по-къс долен крайник, възможна липса на латералната част на глезенната става, срастване на пръстите (синдактилия) или липсата на няколко пръсти. Възможно е да липсва и част от ходилото. Най-често е едностранна липсата на кост, но са описани случаи на двустранна липса. Състоянието се забелязва с помощта на ултразвукова диагностика вътреутробно и се налага ампутация на крайника. Други процедури, които се използват са корекционна остеотомия и удължаване на крайника (апаратура на Илизаров), но са по-скъпи и понякога водят до деформация.
Има описани случаи на липсващ голям пищял (тибия). Отново липсата може да бъде едностранна или двустранна, като по-често е засегнат десният крайник. Състоянието се нарича тибиална хемимелия. Клинично се представя със скъсен крайник с деформации в областта на коляното и глезена. Липсват ставни връзки, които дестабилизират костно-мускулната система на долния крайник, вследствие на което е възможно да липсва четириглавият бедрен мускул. Лечението е хирургично, извършва се ампутация.
Вродени аномалии на метакарпалните кости на ръката се срещат сравнително рядко, но са чести симптоми при различни синдроми. Според класификацията на Риордан се делят на:
- дефект на лъчевата кост (много тежко състояние)
- частичен дефект на лъчевата кост (тежко)
- хипопластична лъчева кост (средна степен)
- нормална лъчева кост (нормално)
 Заедно с дефект на лъчевата кост понякога липсват останали кости, а при някои пациенти и пръсти. Срещат се и пациенти с хипопластична или липсваща лакътна кост.
Заедно с дефект на лъчевата кост понякога липсват останали кости, а при някои пациенти и пръсти. Срещат се и пациенти с хипопластична или липсваща лакътна кост.
Вродената липса на лигамент (ставна връзка) е рядко срещана малформация. За първи път е описана през 1956 г. Липсата на кръстна връзка на коляното е с честота 0,017 на 1000 живородени деца, като може да е самостоятелно или част от синдром. Диагноза се поставя чрез компютърна томография. За реконструкция на ставната връзка се взима част от сухожилието на големия пищял и се ремоделира. При някои пациенти, освен тази находка са установени дислокация на коляното, абнормалии на менискусите и др.
Вродената липса на една от главите на сухожилието на двуглавия мускул на ръката е рядко състояние, среща се едностранно и по-рядко двустранно, самостоятелно или при различни синдромни състояния.
Синдромът на Клипел-Файл (Klippel-Feil) е рядко вродено заболяване, характеризиращо се със сливане на няколко от шийните прешлени, изграждащи гръбначния стълб. Според Файл синдромът може да се класифицира в следните три типа:
- Тип I - срастване на повече шийни прешлени
- Тип II - сливане на един или повече прешлена
- Тип III - наличие на аномалии на гръбначния стълб във връзка с тип I или тип II
Клипел-Файл се среща много рядко, едва един на всеки 4000 души е засегнат в световен мащаб. За диагностициране се използват рентгенография, ядрено-магнитен резонанс, компютърна томография. Този синдром има голям спектър от симптоми, засягащи различни части на тялото. Поради това са необходими и други изследвания, за да се открият допълнителни физически или функционални аномалии или заболявания.
Карпалната срастване (carpal coalition) представлява сливането на две или повече карпални кости, когато те не успеят да се сегментират по време на вътреутробното развитие. Състоянието е описано за първи път през 1779 г. Често е изолирана деформация, но може да е част и от синдром. Най-често се сливат os lunatum и triquetrum. Карпалните съединявания се поделят на:
- Тип 1 - непълно сливане, заедно с псевдоартроза
- Тип 2 - сливане с малка пролука между костите
- Тип 3 - пълно сливане
- Тип 4 - сливане в комбинация с други аномалии
Други вродени аномалии на костно-мускулната система са сливането на кости, като например краниосиностоза. Между костите на черепа се намират 6 шева (сутури), които позволяват на костите да се разместват докато нарастват. Ако някои от шевовете сраснат преждевременно, останалите кости ще се наместят в други посоки и ще доведе до лицево-челюстни малформации.
Болестта на Крабе (Krabbe disease) е дегенеративно заболяване, което се среща рядко. То е сфинголипидоза, унаследяваща се по автозомно-рецесивен път. Честотата му на поява е 1 на всеки 100 000 души. Установено е, че по-често срещано е в Израел. Децата се раждат нормални, симптомите се проявяват по-късно (3-6 годишна възраст). Изразява се с треска, повишена възбудимост, раздразнителност, скованост в крайниците, гърчове, изоставане във физическото и интелектуално развитие, глухота, спазми, атрофия на зрителния нерв и други симптоми. Засега не съществува лечение за болестта Крабе, но е установено, че трансплантация на костен мозък влияе благоприятно и се използва в ранен етап на заболяването. Лечението на болестта е симптоматично и поддържащо. Физикална терапия може да помогне за поддържане или увеличаване на мускулния тонус и движение.
Торакогастросхизис е вродено заболяване, при което гръдната стена не се затваря правилно по протежението на срединната линия на предната част на гръдния кош и корема. Причината за заболяването е неизвестна, но е установена  връзка с тютюнопушене и употреба на алкохол по време на бременността и млада възраст на майката (под 20 години). Близо 60% от децата се раждат преждевременно. След раждането детето ще има малка дупка в гръдната си стена. Някои от органите е възможно да се подават навън.
връзка с тютюнопушене и употреба на алкохол по време на бременността и млада възраст на майката (под 20 години). Близо 60% от децата се раждат преждевременно. След раждането детето ще има малка дупка в гръдната си стена. Някои от органите е възможно да се подават навън.
Анхидротична ектодермална дисплазия или още синдром на Крис-Сименс-Таурин (Christ-Siemens-Touraine syndrome) е една от 150-те ектодермални дисплазии в човешкото тяло. За заболяването са отговорни мутации в гените EDA, EDAR, and EDARADD. Предполага се, че засяга 1 на 17 000 души. Повечето пациенти не могат да се потят (ахидроза), защото имат по-малко потни жлези и те не функционират правилно. Липсват космени фоликули, затова болните са с рядка коса (хипотрихоза), зъбите им са с малформации или липсват (хиподонтия). Пациентите със синдром на Крис-Симонс-Таурин имат отличителни лицеви черти — изпъкнало чело, плътни устни, плосък мост на носа, кожата е тънка, набръчкана, тъмна около зоната на очите. Пациентите са с хронични кожни проблеми като екзема.
Симптоми и признаци при Други вродени аномалии на костно-мускулната система МКБ Q79.8
ВсичкиБиблиография
https://en.wikipedia.org/wiki/Carpal_coalition
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3341811/
https://gradestack.com/Dr-Bhatia-Medical/Pediatric-Orthopedics/Congenital-Absence-of/16060-3176-59558-study-wtw
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4033818/
https://www.jaapos.org/article/S1091-8531(99)70059-5/abstract
https://boneandspine.com/congenital-absence-of-tibia/
http://medind.nic.in/icb/t09/i11/icbt09i11p1178.pdf
https://www.tandfonline.com/doi/pdf/10.3109/17453677708994803
https://ghr.nlm.nih.gov/condition/klippel-feil-syndrome
https://en.wikipedia.org/wiki/Krabbe_disease
Коментари към Други вродени аномалии на костно-мускулната система МКБ Q79.8