Синдром на Klippel-Feil МКБ Q76.1
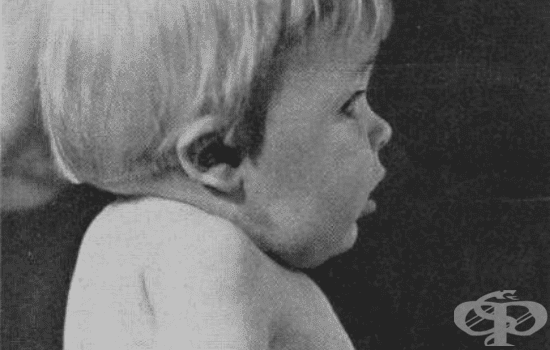
 Рядко вродено заболяване, характеризиращо се със сливане на няколко от шийните прешлени, изграждащи гръбначния стълб е синдромът на Клипел-Файл. През 1912 г. Морис Клипел и Андре Файл самостоятелно предоставят първите описания на синдрома при пациенти със следните прояви:
Рядко вродено заболяване, характеризиращо се със сливане на няколко от шийните прешлени, изграждащи гръбначния стълб е синдромът на Клипел-Файл. През 1912 г. Морис Клипел и Андре Файл самостоятелно предоставят първите описания на синдрома при пациенти със следните прояви:
- Къс и изпъкнал врат
- Намалена подвижност в областта на шията
- Ниска линия на косата на задната част от главата
Повечето засегнати хора имат една или две от тези характерни особености. По-малко от половината от всички пациенти със синдром на Клипел-Файл имат всичките три класически признака на това състояние.
Според Файл синдромът може да се класифицира в следните три типа:
- Тип I - Масивно срастване на шийни прешлени
- Тип II - Сливане на един или два прешлена
- Тип III - Наличие на аномалии на гръбначния стълб във връзка с тип I или тип II на синдрома
Пациентите със синдром на Клипел-Файл обикновено манифестират болестта в ранно детство, но понякога тези признаци се появяват по-късно в живота. Предизвикателството пред лекаря е да разпознае свързаните със синдрома аномалии и да предприеме най-правилните действия.
Този синдром може да причини хронично главоболие, както и болка в областта на шията и гърба. Понякога синдромът на Клипел-Файл се среща като признак на друго разстройство или аномалия. Пример за това са синдромът на Вилдерванк (Wildervanck syndrome, характеризиращ се с парализа на шести черепно-мозъчен нерв (n. abducens), увреждане на слуха, къса шия, сраствания на прешлени и др.) или хемифациална микрозомия (асиметрична лицево-челюстна малформация).
В тези случаи пациентите имат характеристиките както на синдрома на Клипел-Файл, така и на допълнителното разстройство.
Клипел-Фейл се среща много рядко, едва при 1 на 40000 деца в световен мащаб. Предполага се, че синдромът се причинява от мутации в гени GDF6, GDF3 или MEOX1, като те са отговорни за произвеждането на белтъци в тялото, подпомагащи развитието на костите и правилното им сформиране. Белтъкът, произведен от гена GDF6, е необходим за правилното образуване на гръбначния стълб, както и на други кости и стави. Протеинът, произведен от гена GDF3 също участва в развитието на костите, но неговата точна роля е неуточнена.
Генът MEOX1 е отговорен за производството на т.нар. homeobox МОХ-1 протеин, който регулира процеса на отделяне и диференциране на прешлените един от друг, по време на ембрионалното развитие. Това се налага, тъй като преди това те са слети заедно, формирайки гръбначния стълб.
Мутациите в гените GDF6 и GDF3, вероятно водят до намалена функция и производство на съответните протеини, което води до появата на синдрома на Клипел-Файл. МЕОХ1 генните мутации също водят до пълна липса на homeobox МОХ-1 протеин. Въпреки че GDF6, GDF3 и homeobox MOX-1 протеините участват в развитието на костите, не е ясно как недостига на някой от тях води до непълно разделяне на шийните прешлени при хора със синдром на Клипел-Файл.
 В някои случаи синдромът на Клипел-Файл изглежда се поражда случайно или по неизвестни причини (идиопатично). В други случаи аномалията се унаследява по автозомно-доминантен или автозомно-рецесивен модел и е възможно да се наблюдава в повече от един член на семейството.
В някои случаи синдромът на Клипел-Файл изглежда се поражда случайно или по неизвестни причини (идиопатично). В други случаи аномалията се унаследява по автозомно-доминантен или автозомно-рецесивен модел и е възможно да се наблюдава в повече от един член на семейството.
Автозомно-доминираща е аномалията, когато се наблюдава мутация в гените GDF6 и GDF3. Това означава, че едно копие на мутиралия ген във всяка клетка е достатъчно, за да предизвика аномалията.
Когато човек с автозомно-доминантно състояние има деца, всяко дете има 50% шанс да наследи мутиралото копие на гена.
Ако синдромът се причинява от мутации в гена MEOX1, той се унаследява по автозомно-рецесивен модел. Това означава, че индивидът трябва да унаследи две променени копия на мутиралия ген във всяка клетка, за да бъде засегнат. Родителите на индивид с автозомно-рецесивно заболяване носят по едно копие на мутиралия ген, но те не са засегнати.
Когато двама здрави носители на синдрома на Клипел-Файл с автозомно-рецесивно предаване имат деца, всяко дете има 25% риск да бъде засегнато, 50% шанс да бъде незасегнат преносител (като всеки от родителите си) и 25% шанс да бъде незасегнато и да не бъде преносител.
Синдромът на Клипел-Файл обикновено се диагностицира след извършването на рентгенологично изследване или друг образно-диагностичен метод. След като се открие сливането на шийните прешлени, се прави рентгенологично изследване на целия гръбначен стълб, за да се открият други гръбначни аномалии. Възможно е да са необходими допълнителни изследвания като компютър-томографски или ядрено-магнитни, за да се оцени степента на аномалията и евентуално засягане на меките тъкани.
Този синдром може да се свърже с широк спектър от аномалии, включващи много и различни части на тялото. Поради това са необходими и други изследвания, за да се открият допълнителни физически или функционални аномалии или заболявания.
Изследванията включват:
- преглед на гръдния кош, за да се изключат увреждания на сърцето и белите дробове
- изследване на гръдната стена за откриване на възможни аномалии на ребрата
- ЯМР за стесняване на гръбначно-мозъчния канал или неврологични дефицити
- ехографско изследване на бъбреците за бъбречни аномалии
- оценка на слуха поради високата честота на загуба на слух
- различни лабораторни тестове за оценка на функциите на органите
 За съжаление няма лечение за синдрома. Терапията обикновено е насочена срещу симптомите и поддържа благосъстоянието на засегнатия пациент. Терапията е индивидуална и зависи от характеристиките и тежестта на аномалията при всеки човек, като е възможно то да продължи през целия живот.
За съжаление няма лечение за синдрома. Терапията обикновено е насочена срещу симптомите и поддържа благосъстоянието на засегнатия пациент. Терапията е индивидуална и зависи от характеристиките и тежестта на аномалията при всеки човек, като е възможно то да продължи през целия живот.
Необходими са внимателна оценка, диагностика, правилна терапия и координация с различни специалисти, за да се подобри състоянието и да се гарантира, че няма да се пропусне увреждане.
Съществуват различни консервативни терапии, включително използването на шийни яки, физиотерапия, нестероидни противовъзпалителни средства (НСПВС) и различни лекарства за облекчаване на болката. Въпреки това, при много хора със синдром на Клипел-Файл, симптомите прогресират поради дегенеративни промени, които се появяват в областта на гръбначния стълб.
Хирургическите интервенции могат да бъдат наложителни при:
- постоянна болка в областта на гръбначния стълб
- нарушение в неврологичната дейност
- гръбначна нестабилност
- свиване на гръбначно-мозъчния канал
- коригиране на тежка сколиоза
Някои хора с Клипел-Файл може да се нуждаят от операция за лечение на други скелетни аномалии или такива, свързани със:
- сърцето
- бъбреците
- ушите
- очите
- други части на тялото
Тези с повишен риск от неврологични усложнения или заболявания трябва редовно да се наблюдават от лекар и близките си, като е препоръчително да избягват дейности, които могат да доведат до травми или нараняване на шийните прешлени.
Дългосрочната прогноза за хората със синдром на Клипел-Файл варира в зависимост от специфичните характеристики и степента на засягане на всеки човек. Докато всички пациенти имат сливане на поне два шийни прешлена, евентуалните допълнителни признаци и симптоми могат да се различават значително.
По принцип хората с минимални или слабо изявени аномалии могат да водят нормален, активен живот и да нямат значителни ограничения или затруднения. Хората с допълнителни аномалии и/или тежки форми на заболяването може да изискват внимателно проследяване и лечение, но да имат добра прогноза при правилно лечение.
Симптоми и признаци при Синдром на Klippel-Feil МКБ Q76.1
ВсичкиЛечение на Синдром на Klippel-Feil МКБ Q76.1
Библиография
https://ghr.nlm.nih.gov/condition/klippel-feil-syndrome
https://emedicine.medscape.com/article/1264848-overview
https://www.chop.edu/conditions-diseases/klippel-feil-syndrome
https://rarediseases.info.nih.gov/diseases/10280/klippel-feil-syndrome
https://radiopaedia.org/articles/klippel-feil-syndrome-3
Коментари към Синдром на Klippel-Feil МКБ Q76.1