Синдром на Ehlers-Danlos МКБ Q79.6
Синдром на Ehlers-Danlos (EDS, Елерс-Данлос) представлява група от наследствени системни заболявания с нарушения на съединителната тъкан, дължащи се на дефект в синтеза на колаген (тип I или III). Колагенът придава здравина на съединителната тъкан.
Синдром на Ehlers-Danlos (EDS, Елерс-Данлос, СЕД) е известен и като:
- Синдром на Елерс-Данло
- Синдром на Черногубов-Елерс-Данлос
- Синдром на Meekrin-Ehlers-Danlos
- Еластична фибродисплазия
- Генерализирана еластична фибродисплазия
- Вродена множествена ставна слабост
- Хипереластична кожа (Cutis hyperelastica)
- Несъвършенна десмогенеза на Русаков
Според англоезични източници се засягат до 1 на 5000 души. Първоначално оценките за разпространението са варирали от 1 на 250 000 до 1 на 500 000 души, но скорошните проучвания са доказали много по-голяма честота. Една от най-засегнатите групи се оказват евреите ашкенази, като мутацията тип VIIc Ehlers-Danlos при тях достига до 1 на 248 души.
Причини за възникване на синдрома са мутации в различни генни локуси. В зависимост от степента на мутацията, проявите на заболяването може да са от незначителни до животозастрашаващи. Мутациите водят до промяна в структурата или количеството на колагена или протеините, взаимодействащи с колагена. Болестта се унаследява по-често автозомно-доминантно и по-рядко автозомно-рецисивно. Засегнатите гени и ензими са: COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, TNXB, ADAMTS2, PLOD1, B4GALT7, DSE и D4ST1/CHST14. Понякога генът не се унаследява в потомството, а възниква спонтанно.
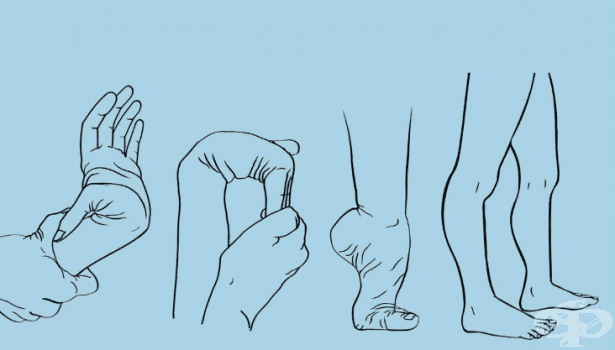
 Болестта поразява ставите, кожата и кръвоносните съдове. Основните прояви на EDS от страна на костно-мускулната система са: ставна халтавост, ставна хипермобилност (подвижност, надвишаваща нормалната за съответната става), изключително лесни ставни луксации (изкълчвания) и сублуксации, болест на Osgood-Schlatter, деформация на пръстите на ръцете като “лебедова шия“, плоско стъпало, тясно и високо небце, остеопения (ниска плътност на костите), изключително ранно започнала остеопороза (от 30-годишна възраст), мускулна слабост, засилваща се при студено време, кифоза (гърбица), сколиоза (странично изкривяване на гръбначния стълб), нестабилност на главата поради слабост на сухожилията на гръбначния стълб, болки в мускулите, лесна уморяемост.
Болестта поразява ставите, кожата и кръвоносните съдове. Основните прояви на EDS от страна на костно-мускулната система са: ставна халтавост, ставна хипермобилност (подвижност, надвишаваща нормалната за съответната става), изключително лесни ставни луксации (изкълчвания) и сублуксации, болест на Osgood-Schlatter, деформация на пръстите на ръцете като “лебедова шия“, плоско стъпало, тясно и високо небце, остеопения (ниска плътност на костите), изключително ранно започнала остеопороза (от 30-годишна възраст), мускулна слабост, засилваща се при студено време, кифоза (гърбица), сколиоза (странично изкривяване на гръбначния стълб), нестабилност на главата поради слабост на сухожилията на гръбначния стълб, болки в мускулите, лесна уморяемост.
Кожата е също засегната, като могат да се появи епикантус (кожна гънка на клепача), лесно да се получат кръвонасядания, трудно зарастващи рани или кожата да е толкова тънка, че да прозира.
От страна на сърдечно-съдовата система се забелязват малки и силно раними кръвоносни съдове, пролапс на митралната клапа, разширени вени, ендокардит, разширяване (аневризма) на възходяща аорта или синдром на ортостатичната тахикардия и тежки аритмии.
Други възможни прояви на синдрома са:
- Малформация на Арнолд-Киари (поражение на продълговатия и малкия мозък с нарушение на гълтането и силни болки в тила)
- Болест на Рейно
- Функционални разстройства на храносмилателната система (гастрит и синдром на раздразненото черво)
- Кървене или перфорация от стомашно-чревния тракт
- Невралгия на нервус тригеминус и парестезия на различни нерви
- При бременност е възможен спонтанен аборт
- Новородените деца са със слаб мускулен тонус
- Малките деца прохождат трудно
- Различни хернии
- Пролапс на ануса
- Симптом на Горлин (докосване на езика до носа)
- Спонтанен пневмоторакс
- Сънна апнея
- Нарушена агрегацията на тромбоцитите
- Късогледство
- Сини склери
- Кървене от венците, тежка пародонтоза, множество кариеси
- Повишен риск от усложнения при хирургични операции
Поради огромното клинично разнообразие на симптоми, класификацията на синдрома на Елерс-Данлос (EDS) е различна в зависимост от ползваните източници, като руско- и англоезичните класификации се отличават значително. Има разлика и в зависимост от времето когато е публикувана дадена класификация. Според рускоезичната литература се счита, че има 10 основни типа, като 6 от тях са с разшифровани хромозомни поражения. Тук ще бъде цитирана и англоезична класификация публикувана през 2017 година. Не за всички форми на синдрома са установени точните генни мутации.
 Руската класификация с установени 6 форми на синдрома е:
Руската класификация с установени 6 форми на синдрома е:
- Ставна хипермобилност е форма на EDS, свързана с мутация в гена COL3A1. Има автозомно-доминантно унаследяване и засягане на малки и големи стави. Налице е ставна хипермобилност и често се диагностицира грешно като фибромиалгия. Характерна е ранно настъпващата, до 30-годишна възраст, остеопороза.
- Класическa форма на EDS - свързана е с мутация в гена COL5A1. Проявява се с хеморагичен синдром, силно разтеглива кожа, формиране на подкожни кисти и доброкачествени кожни тумори (меланообразуващи или епителни).
- Съдова форма на EDS - дължи се на дефектен ген COL3A1 и засягане на колаген тип III. Дефектът се унаследява автозомно-доминантно. Тази форма е животозастрашаваща, поради възможност от разкъсване на кръвоносни съдове и дифузни вътрешни кръвоизливи. Засегнатите лица рядко преживяват над 50 години.
- Ахондропластичен тип EDS - форма на синдрома, свързана с дефект на гена COL1A2 и проявяваща се с вродена луксация на тазобедрената става, ранни прояви на артрити, свръхеластична кожа.
- Дерматоспараксис “Dermatosparaxis“ - форма на синдрома, свързана с дефект на ген ADAMTS2. Характеризира се със свръхранима кожа и трудно заздравяващи рани. При засегнатите лица много рано започват костни и мускулни болки.
- Кифосколиотична форма на EDS - дължи се на автозомно-доминантно унаследен дефект на ген PLOD1 и недостиг на ензима лизин-хидролаза. Характеризира се със слаб мускулен тонус и обездвижване към 20-30 годишна възраст.
Според наскоро публикувана (2017 година) англоезична класификация, синдромът на Ehlers-Danlos (EDS) се дели на следните типове:
- Класическа форма на EDS - налице е изключително еластична (разтеглива), гладка кожа, която е крехка и лесно ранима. Може също да се наблюдават псевдотумори на кожата и кисти, разположени предимно в предмишниците и гърдите). Може да се прояви с мускулна хипотония, миалгия, забавено двигателно развитие и хипермобилност на ставите.
- Съдова форма на EDS - характеризира се с тънка и полупрозрачна кожа, която е изключително крехка и лесно ранима. Артериите и някои органи са склонни към разкъсване. Хората, поразени от този тип EDS, обикновено имат нисък ръст, тънка кожа на скалпа и характерни черти на лицето с големи очи и тънък нос. Съществува хипермобилност на ставите, но обикновено се ограничава до малките стави (пръсти). Други чести аномалии са изкривяване на ходилото и / или разкъсване на мускулите, акрогерия (преждевременно стареене на кожата), ранна проявя на разширени вени, пневмоторакс (колапс на белия дроб) и намалено количество на подкожни мазнини.
- Кифосколиозна форма на EDS - свързана е с тежка мускулна хипотония при раждането, забавено двигателно развитие, прогресивна сколиоза още от самото раждане. Засегнатите лица имат крехки артерии, които са склонни към разкъсване, необичайно малки роговици и остеопения (ниска костна плътност). Други чести черти включват "марфаноиден хабитус", който се характеризира с дълги и тънки пръсти и необичайно дълги крайници, хлътнал гръден кош (pectus excavatum) или изпъкнал гръден кош (pectus carinatum).

- Артрохалазия - форма на EDS, характеризираща се с тежка ставна хипермобилност и вродена деформация на тазобедрената става. Други чести признаци включват крехка и еластична кожа, мускулна хипотония, кифосколиоза (кифоза и сколиоза) и лека остеопения.
- Дерматопраксис (Dermatosparaxis) е форма на EDS, свързана с изключително крехка кожа, водеща до тежки подкожни кръвоизливи, белези и излишна кожа, особено на лицето. Има и синдром на крехката корнея, характеризиращ се с тънка роговица и сини склери. Често се появяват различни по локация хернии.
- Подобна на класическата форма на EDS (clEDS) - характеризира се с кожна хиперразтегливост. Кожата е с кадифена текстура и липса на атрофични белези. Налице е генерализирана хипермобилност на ставите с или без повтарящи се луксации (най-често рамо и глезен) и лесно ранима кожа.
- Спондилодиспластична форма на EDS (spEDS) - характеризира се с нисък ръст и мускулна хипотония.
- Мускулно-контратратурна форма на EDS (mcEDS) - характеризира се с вродени множествени контрактури, характерни морфологични изменения на черепа и кожни признаци с атрофични белези по кожата.
- Миопатична форма на EDS (mEDS) - характеризира се с вродена мускулна хипотония и / или мускулна атрофия. Контурът на проксималните стави (ставите на коляното, бедрената кост и лакътя) е нарушен, има и хипермобилност на дисталните стави на глезените, китките, краката и ръцете.
- Пародонтална форма на EDS (pEDS) - характеризираща се с тежък и неконтролируем парадонтит с ранно начало (детство или юношество), претибиални плаки. Има и фамилна история на роднини от първа степен, която отговарят на клиничните критерии.
- Сърдечно-клапна форма на EDS (cvEDS) - характеризира се с тежки прогресивни сърдечно-клапни проблеми (аортна клапа, митрална клапа), кожни проблеми (свръхразтегливост, атрофични белези, тънка кожа, лесно ранима) и хипермобилност на ставите (генерализирана или ограничена до малките стави).
- Хипермобилен EDS - характеризира се главно с хипермобилност на ставите, засягаща както големи, така и малки стави, което може да доведе до повтарящи се сублуксации (частично разместване на ставната цялост). По принцип хората с този тип EDS имат мека, гладка кадифена кожа и хронични болки в мускулите и / или костите.
Диагностицирането на EDS може да бъде направено чрез оценка на медицинската история и клинично наблюдение. Необходимо е извършването на кожна биопсия, доказваща колагенните генни мутации и с нейна помощ могат да се докажат до 95% от случаите. Изследва се и лизилхидроксилаза или оксидазна активност.
В урината се изследва хидроксилизил пиридинолин (hydroxylysyl pryridinoline), той е показателен за синдрома. Изследва се ставната хипермобилност двустранно - на кутретата и палците на ръцете, лакътните и коленните стави.
Ако има множество засегнати индивиди в едно семейство, може да се извърши пренатална диагностика, като се използва генетично изследване. При бременни от семейство с EDS е препоръчително извършване на амниоцентеза - с цел търсене на генетични мутации на плода. При около 30% от засегнатите лица има подкожни кисти, изглеждащи на рентгенова снимка като калцификати (калциеви отлагания в тъканите).
В диференциално-диагностичен план се обсъждат: синдромът на Марфан, генетичното разстройство на съединителната тъкан, синдромът Loeys-Dietz и други.
Синдромът на Ehlers-Danlos често се пропуска като заболяване и не се обсъжда като евентуална диагноза. Една от причините е неговото многообразие от симптоми, водещи до пропуски в диагностиката.
Не съществува радикално лечение на синдрома на Ehlers-Danlos. Лечението е симптоматично в зависимост от конкретните прояви на заболяването и включва: физиотерапия, витамини и конкретно витамин С, кръвоспиращи, преливане на тромбоцитна маса и други. Наложително е да се използват слънцезащитни кремове при засегнатите от синдрома лица, излагащи се често на слънце. Трябва много да се внимава с пациенти, засегнати от синдрома, при хирургични интервенции. Пациентите трябва да избягват тежките физически натоварвания. В тежките случаи е неизбежна инвалидизацията на пациентите и ограничение в продължителноста на живота.
Симптоми и признаци при Синдром на Ehlers-Danlos МКБ Q79.6
ВсичкиЛечение на Синдром на Ehlers-Danlos МКБ Q79.6
Продукти от Framar.bg

Библиография
http://meduniver.com/Medical/genetika/sindrom_elersa-danlosa.html
https://ru.wikipedia.org/wiki/Синдром_Элерса_
https://en.wikipedia.org/wiki/EhlersDanlos_syndromes
https://rarediseases.info.nih.gov/diseases/6322/ehlers-danlos-syndromes
https://radiomed.ru/publications/sindrom-elersa-danlo-sindrom-0
ФЕДЕРАЛЬНЫЕ КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ ПО ДИАГНОСТИКЕ И ЛЕЧЕНИЮ СИНДРОМА
ЭЛЕРСА-ДАНЛО
Коментари към Синдром на Ehlers-Danlos МКБ Q79.6
Пациент
Статията има нужда от обновяване и съдържа непълна, невярна и неточна информация.
Здравейте! Благодарим Ви за сигнала! Информацията в статията ще бъде прегледана и актуализирана.
Статията има нужда от обновяване и съдържа непълна, невярна и неточна информация.