Други уточнени вродени аномалии на костите на черепа и лицето МКБ Q75.8
Костите съставляват пасивната част на опорно-двигателната система и се развиват от клетки на мезодермата (среден зародишен лист) и нервния гребен от 4-та г.с до 8-a г.с. (гестационна седмица).
Въпреки че плодът е добре защитен, някои фактори от външната среда могат да окажат тератогенен ефект, т.е. да доведат до малформации. Тератогенни фактори са:
- Алкохол
- Тютюнопушене
- Кофеин
- Инфекциозни агенти - рубеола, цитомегаловирус
- Тетрациклини
- Антихипертензивни средства.
- Витамин А (ретинол) и др.
Генетичната предразположеност също играе важна роля.
Липсващи кости може да има във всяка една част от тялото:
В МКБ Q75.8 Други специфични дефекти на черепа и костите на лицето са включени три заболявания:
- Вродена липса на кости
- Вродена деформация на челната кост
- Платибазия.
- Вродена липса на кости на черепа:
 Акранията е рядко вродено заболяване. Среща се при 1:1000 живородени деца. Аномалията се появява през 4 г.с, когато предната част на невралната тръба се затвори и нормалната миграция на мезенхимна тъкан, която трябва да образува черепа, не се осъществи. В такъв случай единствено ектодермата покрива една тънка амниотично подобна мембрана. Така мускулните обвивки не могат да се образуват. Поради липсата на вгъване мозъчната тъкан не може да се диференцира в две полукълба.
Акранията е рядко вродено заболяване. Среща се при 1:1000 живородени деца. Аномалията се появява през 4 г.с, когато предната част на невралната тръба се затвори и нормалната миграция на мезенхимна тъкан, която трябва да образува черепа, не се осъществи. В такъв случай единствено ектодермата покрива една тънка амниотично подобна мембрана. Така мускулните обвивки не могат да се образуват. Поради липсата на вгъване мозъчната тъкан не може да се диференцира в две полукълба.
Акранията излага мозъчната тъкан на контакт с амниотичната течност, като се създава риск от разрив със стената на матката. Тъй като мозъчната тъкан на плода е незащитена, следва дегенерация и деструкция, поради механична и химична травма. Стига се до пълното изчезване на мозъчната тъкан до 14 г.с.
Ултразвуковата диагностика с помощта на ехограф позволява ранното диагностициране на дефекта и е необходимо да се вземат мерки веднага, тъй като дефектът е летален.
 Синдромът на Klippel-Feil (Ostrium Furst) се характеризира с къса шия и намалени движения в шийната област, поради намален брой прешлени или сливане на някои от тях. Дължи се на мутации в гените GDF6,GDF3, MEOX1 и се предава автозомно-рецесивно. Пациентите имат нормално развитие, освен ако пациентът има и други заболявания. Болните се оплакват от болки в тила и главата. При някои може да се забележи асиметрия в двете лицеви половини. След време пациентите могат да развият стеноза (стеснение) на гръбначно-мозъчния канал. При някои се открива и остеоартрит в засегнатата област, промени в сетивността, сколиоза. Наблюдават се и дефекти, свързани със слух, зрение, дишане, анормални бъбреци, яйчници, бели дробове.
Синдромът на Klippel-Feil (Ostrium Furst) се характеризира с къса шия и намалени движения в шийната област, поради намален брой прешлени или сливане на някои от тях. Дължи се на мутации в гените GDF6,GDF3, MEOX1 и се предава автозомно-рецесивно. Пациентите имат нормално развитие, освен ако пациентът има и други заболявания. Болните се оплакват от болки в тила и главата. При някои може да се забележи асиметрия в двете лицеви половини. След време пациентите могат да развият стеноза (стеснение) на гръбначно-мозъчния канал. При някои се открива и остеоартрит в засегнатата област, промени в сетивността, сколиоза. Наблюдават се и дефекти, свързани със слух, зрение, дишане, анормални бъбреци, яйчници, бели дробове.
Диагностицирането се извършва още в пренаталния период чрез ехограф.
Птичи лице или още синдром на Пиер Робин е вродено заболяване, характеризиращо се с лицеви деформации - новороденото има по-малка долна челюст (микрогнатия) от нормалното, език, разположен по-назад (глосоптосис) и отвор в небцето. Тези аномалии водят до проблеми с дишането и храненето на индивида. В 20-40% от случаите болестта се развива самостоятелно, но понякога може да има и други симптоми.
Причините за появата на болестта са делеции (отпадания) или мутации на части от ДНК, или други хромозомни аномалии. Синдромът на Пиер Робин e последователност от генетични промени, защото когато долната челюст изостане в развитието си вътреутробно, въвлича и останалите части. Изолирано заболяването се наблюдава при 1:8,500 до 14,000 души. В повечето случаи, то не е унаследено, защото се е развило от нова мутация.
Диагноза се поставя след направени генетични тестове.
Лечението включва множество хирургични манипулации за подобряване на дишането и коригиране на дефектите.
Краниофенестрия (Краниолакуния) - вроден костен дефект на черепа, при който в някои части липсва костна тъкан, в други е изтънена. Дължи се на абнормална продукция на колаген и неправилна осификация. При образна диагностика се виждат кръгли или овални дупки, наречени лакуни. Проблемът се решава след 6 месечна възраст
Вродените деформации се причиняват от тератогенни фактори, вродени инфекции, хромозомни аномалии, генни мутации, фетално-алкохолен синдром.
Към другите уточнени вродени аномалии на костите на черепа и лицето спадат:
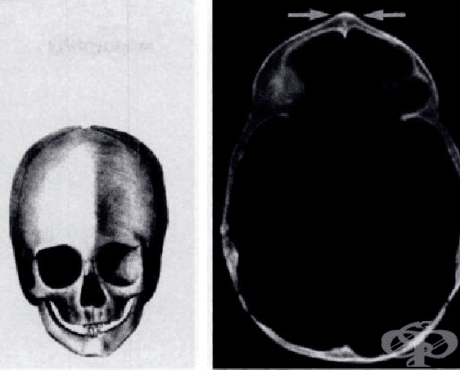
Краниосиностоза (Craniosynostosis) - засегнати са 1 на всеки 2000-2500 живородени деца. Между костите на черепа се намират 6 сутури (шева), това им позволява да се разместват докато нарастват. Ако някои от сутурите срастнат преждевременно, останалите кости ще се наместят в други посоки и ще доведе до лицево-челюстни малформации.
Черепните шевове могат да изчезнат вътреутробно или сред раждането. Когато това се случи вътреутробно, деформациите са по-тежки и по-рано се появява повишено вътречерепно налягане. Ако шевовете изчезнат по-късно, деформациите не са толкова тежки. Като цяло колкото по-късно зараснат шевовете, толкова по-леко протича заболяването. Краниосиностозата може да е придружаващ симптом на други заболявания - болест на Крузон, синдром на Carpenter, синдром на Apert, синдром на Pfeiffer.
Симптомите са обусловени от повишеното вътречерепно налягане — пристъпно главоболие, придружено с гадене и повръщане. Някои сухожилни рефлекси могат да са повишени, а други отслабени, могат дори да се появят някои патологични рефлекси. Възможно е да се появят и психични смущения — повишена раздразнителност, умора, нарушения в паметта. Могат да се появят гърчове. Екзофталмът (изпъкване на очната ябълка) се появява често, може да бъде едностранен или двустранен.
Диагностика се извършва с рентгенографии, компютърна томография, ядрено-магнитен резонанс, изследване на ликвора.
Лечението е хирургично, като целта е да се овладее повишеното налягане и да се коригира черепната деформация. Интервенцията е хубаво да се извърши до 1 година на детето, преди шевовете да са сраснали напълно.
Плагиоцефалия (Plagiocephaly) — най-често е резултат от срастване на ламбдоидният шев. Асиметрията е в едната половина на черепа и може да обхване ябълчните кости.
Брахицефалия (Brachycephaly) — срастват двата коронарни шева. Челото е по-широко и високо от нормалното.
Тригоноцефалията - вродено заболяване, резултат от преждевременното сливане на методопския шев, поради което челото е с триъгълна форма. Счита се, че освен генетични, причините могат да бъдат метаболитни или медикаментозни. Наблюдават се отклонения в поведението и невро-психическото развитие — дефицит на внимание, депресия. Лечението е хирургично, като с препоръчва да се извърши преди детето да е навършило 1 година, за по-добри резултати.
Скафоцефалия (Scaphocephaly) — засегнат е сагиталният шев, като предната или задната половина на черепа е уголемена и придава издължен вид на черепната кутия. Скъсена е и дистанцията между двете уши.
Синдромът на Крузон е генетично заболяване, дължащо се на мутация във FGFR2 и FGFR3 гените. Наблюдава се автозомно-доминантно унаследяване, като вероятността детето да се роди болно е 50 %. Наблюдава се преждевременно сливане на някои кости на лицето и това пречи на растежа на черепа и засяга неговата форма и размери, вследствие, на което някои кости изпъкват, най-често това е челната кост. Могат да се забележат и широки, изпъкнали очи, проблеми със зрението, причинени от плитки очни дъна, очи, които не сочат в същата посока (страбизъм), изпъкване на очните ябълки (екзофталм), седловиден нос и недоразвита горна челюст (хипоплазия). Това, което присъства във всеки пациент е черепната синостоза, най-често — брахицефалия, къса и широка глава. Освен това, хората със синдром на Крузон могат да имат и проблеми със зъбите и слуха. Усложнение на заболяването е болест на Мениер (Meniere).
Диагнозата на заболяването се поставя чрез правилно снети анамнеза и физикален преглед, както и генетични тестове, ядрено-магнитен резонанс, компютърна томография, рентгенография.
Налага се хирургично лечение, като целта е да се предотврати срастването на черепните шевове.
Уплътняване на челната кост (Hyperostosis frontalis interna) – рядко заболяване, което често остава недиагностицирано. Промените могат да се видят на рентгенография или компютърна томография. Предполага се, че се унаследява.
- Платибазия (Platybazia):

Нарича се още плоска основа на черепа и може да е вродена аномалия още от вътреутробното развитие или да се появи по-късно поради омекване на костите, оформящи черепната основа, така те са лишени от обичайните си извивки. Това придава асиметрия на черепа. Причината за това е по-широкият ъгъл, който се образува между основата на черепа, предната черепна ямка и кливуса. Това може да доведе до промени и в първия шиен прешлен (атлас). Най-честият симптом е болка в тилната област, но е възможно и поради компресия на продълговатия мозък и коренчетата на нервите, намиращи се там, да доведе до смущения в гълтачния рефлекс.
Диагноза се поставя чрез рентгенография, компютърна томография, ядрено - магнитен резонанс.
Лечение не е необходимо, освен ако не е установена компресия на мозъчни нерви и нервна тъкан, ако това се случи, се налага отодонна резекция, последвана от сливане на задните кости.
Симптоми и признаци при Други уточнени вродени аномалии на костите на черепа и лицето МКБ Q75.8
ВсичкиПродукти от Framar.bg

КОМПЛЕКТ ХИАЛУРОНОВА КИСЕЛИНА СИ ДЖЕЛИ желирани стика 2 кутии * 31
Библиография
“ Ембриология на човека с клинични корелации”- Х. Чучков, Д. Сиврев 2013г
http://www.ppsca.com/skull.htm
https://rarediseases.org/rare-diseases/hyperostosis-frontalis-interna/
https://omedicine.info/bg/sindrom-kruzona-cherepno-litsevoy-dizostoz.html
https://radiopaedia.org/articles/acrania-anencephaly-sequence
https://sonoworld.com/fetus/page.aspx?id=77
https://pirogov.eu/bg/kraniosinostoza_p177.html
http://neurosurgery.ucla.edu/platybasia
https://ghr.nlm.nih.gov/condition/klippel-feil-syndrome#diagnosis
Коментари към Други уточнени вродени аномалии на костите на черепа и лицето МКБ Q75.8