Краниосиностоза МКБ Q75.0
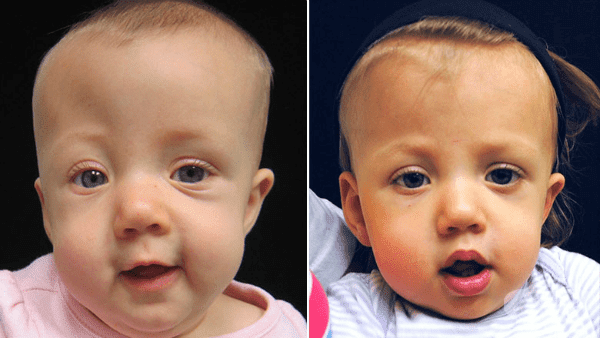
Краниосиностозата е патологично състояние, което се характеризира с преждевременно срастване на костните шевове и нарушаване на пропорционалността между нарастването на мозъчния паренхим и черепната кухина.
Краниосиностозата може да се срещне и под термина краниостеноза. Появява се на всеки 2000 живородени деца, като 15-40% от тях са в комбинация със синдром.
Тъй като мозъкът расте интензивно през първите 2 години от живота е важно костите да не срастват, за да му се даде пространство. Всъщност пълното срастване на костите се случва в юношеските години. Сливането започва през първата година на детето и завършва към 2ра-3та година. Последователността и темпа на срастване определят степента на деформацията. Компенсаторно мозъкът нараства в посоката, в която шевовете не са сраснали все още, с което се променя и неговата форма.
Лекарите не са открили всички причини за появата на заболяването, но засега е установено, че в повечето случаи е генетично.
Краниосиностозата бива:
- Несиндромна — дължи се на мутация на гена EFNA-4. Предполага се, че мутират и FGFRs-1, — 2, — 3 и TWIST-1.
- Синдромна — съществуват над 150 синдрома свързани с краниосиностоза (синдром на Пфайфър, синдром на Крузон, синдром на Карпентър, синдром на Аперт, синдром на Джаксън Уейс и др.).
Някои хематологични състояния могат да доведат до появата на краниосиностоза — таласемия, жълтеница у новороденото, сърповидно-клетъчна анемия. Лекарства с доказано тератогенно действие (увреждащи плода) са тетрациклини, флуконазол, ретиноиди, валпроат и др. Други заболявания, водещи до краниосиностоза са мукополизахаридози тип 1 и 2, хипертироидизъм, рахит, малформации (енцефалоцеле, микроцефалия, холопрозенцефалия).
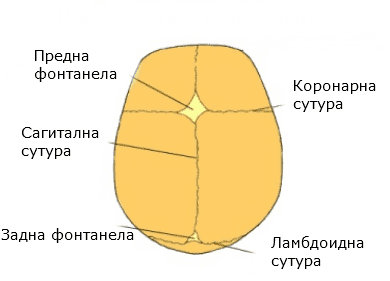
Лесно се разпознава пациент с краниосиностоза, поради особеностите във външния вид. Има значение коя сутура (шев) е срастнала преждевременно, тъй като тя определя накъде компенсаторно е нараснала главата. Поради преждевременното срастване на шевове могат да се получат следните промени в черепа:
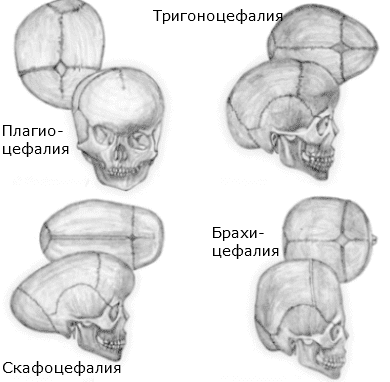 Акроцефалия – вродена деформация на черепа (вид оксицефалия), като главата е с конична форма с продълговат череп и изпъкнало чело. Дължи се на ранно срастване на шева между челната и теменните кости.
Акроцефалия – вродена деформация на черепа (вид оксицефалия), като главата е с конична форма с продълговат череп и изпъкнало чело. Дължи се на ранно срастване на шева между челната и теменните кости.
Брахицефалия – срастват двата коронарни шева. Челото е по-широко и високо от нормалното.
Тригоноцефалия – вродено заболяване, резултат от преждевременното сливане на методопския шев, поради което челото е с триъгълна форма. Счита се, че освен генетични, причините могат да бъдат метаболитни или медикаментозни. Наблюдават се отклонения в поведението и невро-психическото развитие – дефицит на внимание, депресия. Лечението е хирургично, като с препоръчва да се извърши преди детето да е навършило 1 година, за по-добри резултати.
Скафоцефалия (Scaphocephaly) – засегнат е сагиталният шев, като предната или задната половина на черепа е уголемена и придава издължен вид на черепната кутия. Скъсена е и дистанцията между двете уши.
В зависимост от това кой шев е засегнат се подразделят на:
- Сагитална синостоза 40-45%
- коронарна синостоза 20-25%
- Метопична 5-15%
- Множествена синостоза 5-15%
- Ламбдоидна синостоза 0-5%
Сагиталната синостоза е най-често срещания вариант, като в повечето случаи не е част от синдром. Компенсаторно нараства черепният покрив (калвария), придавайки издължен, скафоцефаличен вид на главата.
Коронарната синостоза може да бъде едностранна или двустранна, като при едностранната се наблюдава сплескване на челото само от едната страна. Освен това води до невъзможност на едното крило на сфеноидалната кост да се спусне надолу, причинявайки т, нар. "Артилерийска" деформация с издължаване на орбитите на очите. Двустранната синостоза води до скъсяване на предно — задния размер на черепа и брахицефалия.
Метопичната синостоза се свързва с тригоноцефалия, оформя се триъгълен череп и се скъсява разстоянието между очите. Теменната кост компенсаторно се разширява и удължава.
Ламбоидната синостоза се среща рядко и клинично трудно се различава от позиционна плазиопатия. Заради нея компенсаторно едностранно се сплесква тилната кост и мастоидната кост изпъква. Понякога контралатерално се забелязва разрастване на теменната кост и основата на черепа се накланя.
 Понякога се случва пациентите да се оплакват от постоянно главоболие, заради нарастващото вътречерепно налягане.
Понякога се случва пациентите да се оплакват от постоянно главоболие, заради нарастващото вътречерепно налягане.
Най-често свързаният с краниосиностоза синдром, е този на Крузон. Проявява се под формата на брахицефалия, като сраснат двата коронарни шева и челото е разположено по-високо и широко от нормалното. По-рядко се наблюдава акроцефалия (заострен череп), тригоноцефалия (челото е с триъгълна форма), плагиоцефалия (сплескана глава), платибазия (плоска основа на черепа). Появява се на 25 000 живородени деца. Около 30% от пациентите имат и хидроцефалия.
Синдромът на Аперт също се характеризира с билатерална коронарна краниосиностоза с издължено чело, аномалии в средната част на лицето, клюновиден нос, полегати очи, хипертелоризъм. Много наподобява синдрома на Крузон. Той дори по-рядко срещан, на 100 000 живородени деца. Допълнително при някои пациенти присъстват аномалии в ръцете, таза, лактите и коленете. Интелектуалното развитие е засегнато, като е забелязано забавяне в 50-85% от случаите.
Синдромът на Карпентър се дължи на ламбдоидна или сагитална синостоза с аномалии в крайниците, например допълнителни пръсти. Причината не е известна, но заболяването уврежда и сърцето и зъбите. Среща се изключително рядко, едва един на милион души са засегнати.
Синдромът на Пфайфър е също рядко генетично заболяване, засягащо 1:100 000 души по света. Пациентите са с голямо чело, хипертелоризъм, полегати очи, екзофталм (изпъкнали очи), недоразвита горна челюст. Около половината от болните са със загуба на слуха и дентални проблеми.
 Невинаги различна форма на черепа означава, че детето е с краниосиностоза. Ако тилът е плосък вероятна причина е фактът, че детето лежи на едната си страна прекалено дълго време. Деформацията се оправя лесно, като се промени позицията на лежане или се носи специална каска.
Невинаги различна форма на черепа означава, че детето е с краниосиностоза. Ако тилът е плосък вероятна причина е фактът, че детето лежи на едната си страна прекалено дълго време. Деформацията се оправя лесно, като се промени позицията на лежане или се носи специална каска.
Ако не се лекува състоянието се усложнява. Деформациите на черепа ще останат перманентни, ниско самочувствие и социална изолация. Нелекуваното повишено вътречерепно налягане води до усложнения като закъснение в развитието, летаргия (липса на енергия или интерес), гърчове, слепота и други нарушения в зрението.
За да се постови точна и ясна диагноза е необходимо детето да се прегледа от педиатър, неврохирург или пластичен хирург. Физикалният преглед цели да провери дали са налице неравности, ръбове или други аномалии по черепа. За по-сигурно се прави компютърна томография или рентгенография. Ако има съмнения за вродено заболяване се правят генетични тестове.
Леките случаи нямат нужда от хирургична намеса, но лекарят може да предпише специална каска, която да ремоделира главата на детето. За съжаление при повечето пациенти се налага операция. Времето на извършване зависи от типа краниосиностоза. Целта на операцията е да коригира лицевите деформации, да премахне повишеното вътречерепно налягане и последиците от него.
Ендоскопският метод е подходящ за бебета под 6 месечна възраст, при които е обхванат само един шев. Минимално инвазивна процедура е. През малки разрези в черепа се вкарва ендоскоп и се отваря сутурата, позволявайки на мозъка да расте нормално.
Традиционният подход с отваряне на черепната кутия се използва за лица над 6 месечна възраст. Правят се разрези и костите се наместват.
Симптоми и признаци при Краниосиностоза МКБ Q75.0
ВсичкиЛечение на Краниосиностоза МКБ Q75.0
Библиография
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3562251/
https://www.cincinnatichildrens.org/health/c/craniosynostosis
https://en.wikipedia.org/wiki/Craniosynostosis
https://www.mayoclinic.org/diseases-conditions/craniosynostosis/symptoms-causes/syc-20354513
Коментари към Краниосиностоза МКБ Q75.0