Мукополизахаридоза, тип I МКБ E76.0
Мукополизахаридоза, тип I е сравнително рядко срещано заболяване, което се проявява в детска възраст.
Мукополизахаридоза, тип I включва синдромите Hurler, Hurler-Scheie и Scheie.
Мукополизахаридозите са метаболитни нарушения, дължащи се на дефицит на лизозомални ензими, които участват в разграждането на глюкозоаминогликаните.
Глюкозоаминогликаните са важна съставка на екстрацелуларния матрикс, вътреставната течност и съединителната тъкан. Натрупването им в клетките на различни органи нарушава функциите на последните. Мукополизахаридоза тип I е най - често срещания тип.
Патогенезата на заболяването е много специфична. Мукополизахаридоза тип I е рядка, автозомно - рецесивна лизозомна болест на натрупването, причинена от дефицит на лизозомния ензим алфа-L-идуронидаза. В резултат възниква неспособност лизозомите да разградят глюкозоаминогликаните дерматан сулфат и хепаран сулфат. Те се натрупват прогресивно в лизозомите.
Дефицитът на алфа-L-идуронидаза повишава уринната екскреция на дерматан сулфат и хепаран сулфат. Ензимът се кодира от локус в хромозома 4. Различни мутации на гена причиняват различни синдроми - синдром на Hurler, синдром на Scheie или синдром на Hurler - Scheie.
Клинично мукополизахаридоза, тип I е хетерогенно заболяване по отношение на тежестта на симптоматиката.
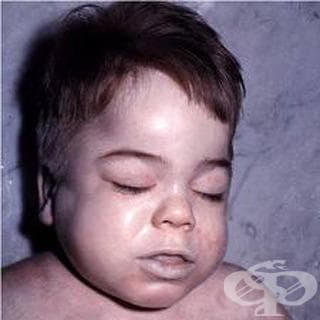
Децата със синдром на Hurler са с нормален вид при раждането, но характерните промени се развиват през първата година от живота. Симптомите включват лицев дисморфизъм, замъгляване на роговицата, хепатомегалия, клапно сърдечно заболяване, обструктивна белодробна болест, изоставане в развитието, загуба на слуха, скелетни деформации и скованост на ставите.
И при трите синдрома Hurler, Hurler-Scheie и Scheie се наблюдава следната кллинична симптоматика.
При пациенти с по - тежката форма на заболяването, типичната симптоматика възниква рано. Продължителността на живот при тях е под 10 години. Пациентите с по - лека форма могат да имат някои от същите симптоми, но са с нормално интелектуално развитие, стоеж и нормална продължителност на живот.
Признаците на лицен дисморфизъм стават видими на възраст 3 - 6 месеца. Главата е широка с изпъкнали челни кости. Коренът на носа стои ниско, върхът е широк с отворени ноздри. Бузите са големи. Устните са уголемени, а устата обикновено остава отворена. Налице е хронична секреция от носа. Замъгляването на роговицата започва през първата година от живота и прогресивно може да доведе до слепота. Среща се също дегенерация на ретината.
При мукополизахаридоза тип I се установяват още прогресивна хепатомегалия, ингвинални и пъпни хернии. От скелетните деформации, които стават видими до 10 - 14 мес. възраст, най - типични са гърбицата и дорзолумбалната кифоза. Тазът е слабо развит с малки феморални глави. На около 2 годишна възраст растежът спира. Характерна е сковаността на ставите с развитие на прогресивна артропатия. Често усложнение е синдромът на карпалния канал.
За диагнозата мукополизахаридоза, тип I основно значение имат повишени нива в урината на дерматан сулфат и хепаран сулфат. Определят се нивата на ензима алфа-L-идуронидаза в куртурирани фибробласти и левкоцити.
Диференциална диагноза се прави с останалите мукополизахаридози.
Пренатална диагностика - измерват се нивата на ензима в амниотичната течност. Допълнителни изследвания - рентгенография, ехокардиография.
Лечението се изразява в ензимозаместващата терапия подобрява белодробната функция, ходенето и намалява натрупването на въглехидрати в органите.
Симптоми и признаци при Мукополизахаридоза, тип I МКБ E76.0
- Ментална ретардация
- Забавен растеж
- Нисък ръст
- Увеличен черен дроб
- Симптоми на челюстта
- Хрема (ринорея)
Лечение на Мукополизахаридоза, тип I МКБ E76.0
Продукти от Framar.bg

Коментари към Мукополизахаридоза, тип I МКБ E76.0