Ранна малкомозъчна атаксия МКБ G11.1
В рубриката ранна малкомозъчна атаксия са разгледани:
- ранна малкомозъчна атаксия с есенциален тремор
- атаксия на Хънт
- ранна малкомозъчна атаксия със запазени сухожилни рефлекси
- атаксия на Фридрайх
- Х-свързана рецесивна спиноцеребеларна атаксия
Атаксия на Хънт
Атаксията на Хънт е рядко дегенеративно неврологично заболяване, характеризиращо се с миоклонус-епилепсия, интенционен тремор, прогресираща атаксия и когнитивно увреждане.
Етиология включва широк спектър от заболявания, причиняващи нарушена връзка между малкия мозък и стволовите ядра.
Началото е в ранна детска възраст и се демонстрира клинично с прогресираща атаксия, интенционен тремор, конвулсии и миоклонични епилептични пристъпи, мускулна слабост, астения, адиадохокинезия и некоординирани движения.
Лечението на атаксията на Хънт е симптоматично.
Х-свързана рецесивна спиноцеребеларна атаксия
Х-свързана рецесивна спиноцеребеларна атаксия включва група от наследствени заболявания, предаващи се чрез Х хромозомата и обединени от общите клинични симптоми на ранна атаксия и други симптоми на малкомозъчно увреждане.
Х-свързана рецесивна спиноцеребеларна атаксия тип 3 се характеризира с ранно начало - още в ранна детска възраст, хипотония, атаксия, невросензорна глухота, изоставане в развитието, езотропия, атрофия на зрителния нерв. Засегнатите пациенти обикновено умират рано, поради кардиопулмонални нарушения.
Х-свързана рецесивна спиноцеребеларна атаксия тип 4 се характеризира с атаксия с начало в ранна детска възраст, засягане на пирамидните пътища, ранна деменция.
Атаксия на Фридрайх
Атаксията на Фридрайх представлява невродегенеративно заболяване с увреждане на спиноцеребеларните трактове, задните стълбци, пирамидните трактове и в по-малка степен - на малкия мозък.
Заболяването се унаследява автозомно-рецесивно. Причинява се от мутация в гена FRDA, който се намира в хромозома 9.
Патоморфология на атаксията на Фридрайх включва лезии, локализирани в гръбначния мозък, дегенерация на задните стълбци, особено в цервикалния отдел, като процесът е по-силно изразен във фасцикулус грацилис. Наблюдава се загуба на по-големите клетки в ганглиите на дорзалните коренчета, дегенерация на пирамидните и задните спиноцеребеларни пътища, особено изразено в лумбалния отдел, загуба на моторни неврони на предните рога, колоната на Кларк, краниалните сетивни ядра, атрофия на мозъчния ствол и горните малкомозъчни крачета, глиоза на бялото вещество на малкия мозък. Церебеларната кора е нормална, въпреки че може да има загуба на клетки на Пуркиние, загуба на дебеломиелинизирани сетивни влакна в периферните нерви, дегенерация на задните коренчета, сетивни ганглии и недостатъчна вторична сегментна демиелинизация.
Други нарушения включват кардиомиопатия с хипертрофия на камерите, дегенерация на кардиомиоцити и интерстициална фиброза.
Патофизиологично се наблюдава мутация в гена FRDA - причинява намаление на протеина фратаксин (frataxin) – желязо свързващ протеин. В резултат на това, в митохондриите се натрупва желязо, което води до токсичност за клетката.
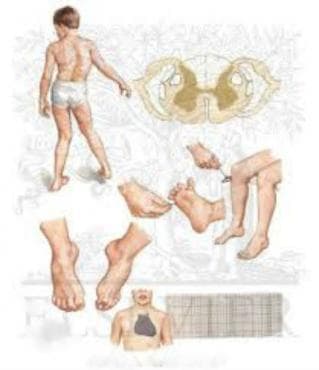
Клиничната картина се характеризира с начало преди 20 години, най-често между 8 и 15 г. Първите симптоми са прогресивна сетивноцеребрална атаксия (нарушена координация) със засягане на походката, тремор на крайниците при покой и при действие, мускулна слабост, дизартрия, нистагъм, намалена зрителна острота, поради атрофия на зрителния нерв, глухота, некоординирано дишане и преглъщане. Установяват се отслабени или липсващи сухожилно-надкостни рефлекси - липсата на коленен и Ахилов рефлекс са много показателни, положителен рефлекс на Бабински. Настъпва увреда на позиционния, дискриминационния и вибрационния усет. Наблюдават се деформации на стъпалото и сколиоза. В допълнение се установява хипертрофична кардиомиопатия, захарен диабет.
При физикалното изследване се установяват:
- церебеларни симптоми - атаксия, нистагъм, дизартрия, дисметрия
- лезия на двигателния неврон - отсъстващи дълбоки сухожилни рефлекси
- пирамидни нарушения – дистална мускулна слабост, удължени или липсващи плантарни рефлекси
- увреда на дорзални стълбци – загуба на вибрационен и проприорецептивен усет
- сърдечни усложнения - кардиомегалия, сърдечни шумове, нарушения в проводимостта
- зрителни нарушения - оптична атрофия, абнормни екстраокуларни движения
- ортопедични нарушения - стъпало на Фридрайх – висок свод, палец - чукче, а останалите пръсти са в дорзална флексия в метатарзофалангеалните стави и в плантарна флексия в интерфалангеалните стави, сколиоза, кифоза
- други - нарушен глюкозен толеранс, нарушена сфинктерна функция (инконтиненция на урината), сензоневронална глухота
Диагностиката на атаксия на Фридрайх започва с преглед от навролог. Електромиографията разкрива дискретни белези на денервация - данни за аксонална сетивна невропатия с привидно нормална скорост по най-бързите двигателни влакна и малки или липсващи сетивни нервни аксонни потенциали. Соматосензорните евокирани потенциали показват забавено централно проводно време. Компютърната томография и ядрено-магнитният резонанс визуализират атрофия на гръбначния мозък, предимно цервикално и леки атрофични церебеларни промени. Продълговатият мозък е с редуциран размер. Ликворът е нормален. Използва се също ЕКГ-диагностика при възникнали сърдечни нарушения.
Критерии на Harding за диагноза атаксия на Фридрайх:
Основни:
- автозомно-рецесивно унаследяване
- начало на симптомите преди 20 годишна възраст
- прогресираща атаксия
- липсващи коленни и Ахилови рефлекси
- малки или липсващи скорости на провеждане на акционните потенциали, отчетена електромиографски
Допълнителни:
- дизартрия
- пирамидна симптоматика на краката
- липсващи рефлекси на ръцете
- дистална загуба на ставномускулния и вибрационен усет в краката
- сколиоза
- патологично ЕКГ
Други:
- нистагъм
- оптична атрофия
- глухота
- дистална слабост и амиотрофия
- пес кавус
- захарен диабет
Диференциална диагноза на ранна малкомозъчна атаксия се прави с наследствена моторна невропатия тип I и други наследствени атаксии.
Липсва специфично етиологично лечение, терапията е симптоматична и включва ортопедични интервенции, използване на ортези и помощни средства, физикална терапия.
Прогнозата е лоша, качеството на живот на пациентите е влошено. Загубата на двигателна способност на долните крайници настъпва средно след 15 години от началото на заболяването при атаксия на Фридрайх, или пациентите не прохождат въобще нормално. Смъртта настъпва средно в третото десетилетие, поради кардиопулмонални усложнения при атаксия на Фридрайх, или в ранна детска възраст при останалите форми на атаксия.
Симптоми и признаци при Ранна малкомозъчна атаксия МКБ G11.1
- Повръщане
- Ментална ретардация
- Усещане за отпадналост
- Гърчове
- Затруднено дишане
- Недостиг на въздух (диспнея)
Коментари към Ранна малкомозъчна атаксия МКБ G11.1