Вродени пороци в развитието на кожата и кожните придатъци
Вродените пороци в развитието на кожата и кожните придатъци се срещат сравнително често. Те могат да бъдат изолирани или да се съчетават с други аномалии като елемент на различни синдроми. По-голямо значение се отдава на разпространените пороци на кожата, които обикновено са причина за смърт на детето.
Кожните вродени аномалии са разнообразна група от заболявания, които са резултат от неправилно развитие на кожата и кожните придатъци. Класификацията на вродени пороци в развитието на кожата и кожните придатъци е следната:
- огнищна аплазия на кожата
- булозна епидермолиза
- ихтиоза
- хиперкератоза
- албинизъм
- невус
- капилярен хемангиом
- синдром на Елерс-Данлос
- хипертрихоза
- атрихоза
- кожни кисти
Булозната епидермолиза е рядка група от наследени заболявания, които се проявяват като мехури или ерозия на кожата, в някои случаи епителната лигавица на други органи, в отговор на малка или невидима травма. Известни са повече от 30 вида булозна епидермолиза, което прави категоризирането на типовете спорно и често объркващо. Международна среща за консенсус във Виена, Австрия през 2008 година отново потвърди следните понастоящем използвани имена за четирите основни вида булозна епидермолиза:
- булозна епидермолиза симплекс - форма на булозна епидермолиза, която причинява мехури на мястото на триене. Обикновено засяга ръцете и краката и се унаследява по автозомно доминантен начин, засягайки кератинните гени.
- свързваща епидермална булоза - наследствена болест, засягаща ламинин и колаген. Това заболяване се характеризира с образуване на блистери в рамките на ламината lucida на зоната на основната мембрана и се унаследява автозомно рецесивно. Също така се представя с мехури на мястото на триене, особено на ръцете и краката, и има варианти, които могат да се появят при деца и възрастни.
- дистрофична епидермолизна булоза - е наследствен вариант, засягащ кожата и другите органи. "Пеперудени деца" е терминът, даден на родените с болестта, тъй като кожата им е толкова деликатна и крехка, колкото крилата на пеперудата. Дистрофичната форма се причинява от генетични дефекти (или мутации) в човешкия COL7A1 ген, кодиращ протеинът на колаген тип VII.
- синдром на Kindler - е рядък подтип на булозна епидермолиза. Синдромът на Kindler е автозомна рецесивна генодерматоза, характеризираща се с вродено акрално образуване на мехури, фоточувствителност, прогресивна пойкилодерма и дифузна кожна атрофия.
Тези основни типове се основават на точното ултраструктурно ниво, при което се получава разделянето, отговорно за появата на мехури. Трите основни типа булозна епидермолиза са клинично и хистологично очертани до 60-те години на миналия век. През 70- те години електронната микроскопия разкрива анормални епидермални кератинови филаменти в булозна епидермолиза симплекс, неразпределените кожни фиксиращи фибрили при дистрофична булозна епидермолиза и дефектни хемидесмозоми в свързваща епидермална булоза.
Цитолизата причинява мехури в епидермиса или в зоната на основната мембрана на кожата. В булозна епидермолиза симплекс, цитолизата причинява мехури в основния или спинозния слой на епидермиса, а кератиноцитите често имат анормална плътност и организиране на кератинови филаменти. В свързваща епидермална булоза, епидермиса се отделя от основната ламината, образувайки блистерна кухина в равнината на ламината lucida, където структурата и плътността на хемамидмозомите често се намаляват.  При дистрофичната епидермална булоза основната ламина остава прикрепена към епидермиса, но блистерната кухина се образува под пластината на дермоепидермалното свързване и фиксираните фибрили могат да изглеждат необичайни, намалени на брой или изобщо липсващи.
При дистрофичната епидермална булоза основната ламина остава прикрепена към епидермиса, но блистерната кухина се образува под пластината на дермоепидермалното свързване и фиксираните фибрили могат да изглеждат необичайни, намалени на брой или изобщо липсващи.
Електронната микроскопия определя нивото на разцепване на кожата при булозна епидермолиза и позволява визуализация и полуколичествена оценка на специфични структури, за които е известно, че са променени при избрани подтипове на заболяването. Наблюдават се някои перинуклеарни отоци и субядрена цитолиза на базалните клетки, но органелите обикновено са непокътнати (фигура 1). Цитолизата се предхожда от агрегиране и натрупване на тонифиламентите, които са прикрепени към хемидесмозомите в дермоепидермалното свързване. Основната мембранна зона е непокътната във всички варианти на булозна епидермолиза симплекс.
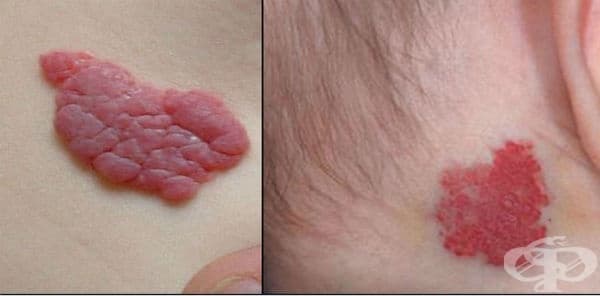
Хемангиомите са доброкачествени тумори, произхождащи от ендотелните клетки, които покриват кръвоносни съдове. Те могат да бъдат капилярни или кавернозни. При капилярните хемангиоми туморът се състои от малки кръвонапълнени места, има розово-червен цвят и прилича на ягода (заглавна снимка). Капилярните хемангиоми растат бързо през първите 1-2 години от живота, а по-късно те стават по-малки и накрая изчезват в 90-95% от случаите. Те са най-често разположени на кожата, и най-вече в областта на главата и шията. Капилярните хемангиоми не изискват никакво лечение в повечето случаи.
Капилярните хемангиоми, спадащи към вродени пороци в развитието на кожата и кожните придатъци, са един от най-честите доброкачествени орбитални тумори в ранна детска възраст. Те са доброкачествени ендотелиални клетъчни неоплазми, които обикновено отсъстват при раждането и характерно имат бърз растеж в детска възраст със спонтанна инволюция по-късно в живота. Това е в контраст с друга известна група детски съдови аномалии, съдови малформации. Съдови малформации, като лимфангиоми и артериовенозни малформации, са налице при раждането и се характеризират с много бавен растеж с постоянство в живота на възрастните.
Предполага се, че капилярните хемангиоми са хамартоматозни пролиферации на съдови ендотелиални клетки. Понастоящем се смята, че те са от плацентен произход поради уникалния микроваскуларен фенотип, който се споделя от младите хемангиоми и човешката плацента. Периорбиталните капилярни хемангиоми следват подобен ход на хемангиомите в други части на тялото. Те обикновено проявяват 2 фази на растеж, пролиферативна фаза и фаза на инволюция. Пролиферативната фаза на бърз растеж обикновено възниква от 8-18 месеца. Патологично се характеризира с увеличен брой ендотелиални и мастоцитни клетки, като последния е стимул за растежа на съдовете. Пролиферацията на ендотелиалните клетки се връща в нормално състояние след фазата на пролиферация.
Инволюционната фаза се характеризира с бавна регресия на хемангиомите. Половината от всички лезии ще инволюират на 5-годишна възраст, а 75% ще инволюират на възраст до 7 години. По време на тази фаза броят на мастните клетки намалява до нормално ниво и се наблюдава намаляване на активността на ендотелиите и мастните клетки. Тези васкуларни пространства се покриват от ендотелиални клетки без наличието на мускулна тъкан.
Рутинната хистопатология варира в зависимост от етапа на хемангиома. При ранната пролиферация, хемангиомите се характеризират с некапсулирани маси и плътни корди от митотично активни ендотелиални клетки в тясна връзка с перицити. Специалните оцветявания разкриват добре развити междинни мембрани около примитивните съдове (фигура 2). Мастните клетки се намират в различен брой във всички етапи. Тъй като хемангиомът се размножава, увеличава се васкулатурата. Увеличаването на апоптотичните ендотелиални клетки и намаляването на митотично активните ендотелиални клетки обявяват фаза на инволюция.
Тъй като инволюцията прогресира, ендотелните клетки продължават да зреят и поемат по-плосък външен вид. Васкулатурата продължава да се увеличава, докато останат малко зрели ектатични съдове. Пролифериращата ендотелиална клетъчна маса може да бъде заменена с фибромастна тъкан. Различните степени на епидермална атрофия, белези и загуба на еластична тъкан могат да се видят при късни инволюиращи лезии.
Пробите могат да бъдат оценени за тъканно-специфични имунохистохимични маркери като GLUT-1, мерозин, Fc-гама-RII и Lewis Y антигени. Тези маркери могат да помогнат за диференциране на инфантилни хемангиоми (положително оцветяване за всички) от други съдови неоплазми или малформации, като например вродени хемангиоми, капосиформен хемангиоендотелиом, или пиогенен гранулом, никоя, от които не оцветява положително за тези антигени. Тези маркери се експресират съвместно от инфантилни хемангиоми, еритроцитни клетъчни мембрани и плацентарни микросъдове.


Продукти свързани със СТАТИЯТА
СУОНСЪН БИОТИН капсули 5000 мкг * 30

ОЛИМП СПОРТ НУТРИШЪН БЕТА-СОЛАР капсули * 30

КОКОНА ФУУТ ГАРД ДЕЗОДОРАНТ ПУДРА ЗА КРАКА 50 г

АКНЕ АУТ АКТИВЕН КРЕМ 30 мл

САПУН БЕБО С ЕКСТРАКТ ОТ СМРАДЛИКА 60 г НИЯ МИЛВА

ТОПИКРЕМ UR-10 ПОДХРАНВАЩ КРЕМ ЗА ТЯЛО ЗА МНОГО СУХА КОЖА С 10% УРЕЯ 200 мл.
Библиография
Color atlas of pathology, Section Congenital malformations
https://emedicine.medscape.com/article/909549-overview#a5
https://emedicine.medscape.com/article/909549-workup#c7
https://emedicine.medscape.com/article/1218805-overview#a6
https://emedicine.medscape.com/article/1083849-workup#c7
СТАТИЯТА е свързана към
- Обща патология
- Смущения в развитието на организма
- Компрес с риванол
- 13 кожни образувания, които е добре да се прегледат от специалист
- Каламинов лосион
- Липа
- Алтернативни средства за лечение на атопичен дерматит
- Червена мъртва коприва
- Алтернативни методи и средства за терапия и контрол на уртикария
- д-р Марияна Иванова Георгиева
- д-р Евдокия Колева Николова
- Алтернативно лечение при изгаряне на кожата
Коментари към Вродени пороци в развитието на кожата и кожните придатъци