Дифузна склероза МКБ G37.0
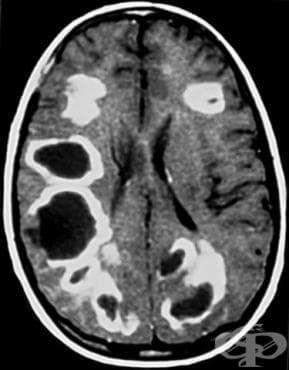
Болестта на Schilder, известна още като дифузна миелинокластична склероза, е много рядко невродегенеративно заболяване, което се проявява клинично като псевдотуморни демиелинизиращи лезии, което затруднява диагнозата.
Обикновено започва в детска възраст, засягайки деца между 5 и 14 години, но са възможни случаи и при възрастни
Първоначалните три случая, описани от Шилдер през 1912, 1913 и 1924 година и означени като encephalitis periaxialis diffusa, впоследствие се оказват три различни състояния. Случаят от 1912 г. е миелинокластична дифузна склероза, случаят от 1913 г. е левкодистрофия, а случаят от 1924 г. е подостър склерозиращ паненцефалит.
Първоначалният случай се появява при 14-годишно момиче, което е било здраво преди развитието на едем на папилата, дясна хемипареза и повишено вътречерепно налягане. Неврологичното й заболяване последва курс от 4-5 месеца на влошаване, водещо до смърт и последващо изследване на мозъка й от Schilder.
Класическата болест на Schilder се счита за една от граничните форми на остра множествена склероза (МС), която се проявява почти изцяло в детска възраст.
Малки, подобни на МС лезии, са описани в някои случаи, придружаващи големите области на демиелинизация.
Други заболявания в тази група са невромиелит оптика, концентрична склероза на Balo и множествена склероза на Marburg.
История
За първи път заболяването е описано от Paul Ferdinand Schilder през 1912 година като фулминантно заболяване на деца с дифузно засегнато бяло мозъчно вещество. В продължение на близо сто години терминът „болест на Шилдер“ е използван за описанието му, но същото име се използва и за някои други патологии на бялото вещество, описани от него.
По-късните анализи са идентифицирали 3 различни заболявания, попадащи под този епоним: адренолевкодистрофия, подостър склерозиращ паненцефалит и истинска болест на Schilder.
През 1986 г. Poser се опитва да ограничи използването на името на болестта на Шилдер, но този термин все още остава двусмислен.
Названието на болестта идва от традиционната класификация на демиелинизиращите заболявания в две групи: демиелинизиращи миелинокластични заболявания и демиелинизиращи левкодистрофични заболявания.
В първата група нормалният и здрав миелин се разрушава от токсично, химическо или автоимунно вещество. Във втората група миелинът е анормален и дегенерира. Втората група е наречена дисмиелинизиращи заболявания от Poser.
Епидемиология
До момента са докладвани по-малко от 20 спорадични случая (предимно мъже).
Преди възможност за ЯМР диагнозата се поставя само при постмортален анализ. Смъртта в докладваните случаи е настъпила в рамките на дни до няколко месеца.
Седем от 9-те случая на болестта на Schilder са настъпили при момчета, всички от които са били на възраст под 10 години. Два допълнителни случая са настъпили при юноши или възрастни жени.
Строгите критерии за болестта на Schilder (т.е. двустранни големи лезии без допълнителни плаки или други находки) водят до 7 пациенти, всички момчета, чието заболяване се е появило главно между 7 и 10-годишна възраст. Два допълнителни случая се наблюдават при жени на възраст над 13 години.
Етиология
Причините за болестта на Schilder са неизвестни. Има доказателства за възможно инфекциозно заболяване в началото на предполагаемата болест на Шилдер.
В някои доклади съществува латентност между това първоначално фебрилно заболяване и субакутното начало на болестта на Schilder. Някои от тези случаи могат да бъдат примери за остър дисеминиран енцефаломиелит.
Други случаи имат фулминантно протичане без такова ясно разграничение между продромалното състояние и началото на болестния процес, смятан за болест на Schilder. Много от тези случаи може да са примери за енцефалит или някакво метаболитно разстройство.
Патофизиология
Патологията на болестта на Schilder е характерна. Широко разпространена демиелинизация на двете церебрални хемисфери с различна степен на увреждане на аксона се открива при аутопсии. Лезиите обикновено са донякъде асиметрични, имат остри граници. Подобни демиелинативни промени често се откриват в мозъчния ствол и малкия мозък.
Увреждането на аксона може да възникне в цялата нервна система, но промените са особено изразени в гръбначния мозък. В тежки случаи патологията по този начин прилича повече на тази на множествената склероза, отколкото на острия дисеминиран енцефаломиелит.
Описни са случаи с характерни двустранни лезии, които са малко по-малки оти, които са свързани с допълнителни множество малки плаки. Появата на тези плаки наподобява обичайната плака при множествена склероза, отколкото типичните остри дисеминирани енцефаломиелитни лезии. Тези случаи обикновено се проявяват в юношеска или зряла възраст, а не в детството, и техният клиничен ход е по-променлив от острите тежки и по-широко разпространени случаи.
Клинична картина
Началото на заболяването обикновено е подостро, въпреки че в някои случаи началото е по-рязко и възниква в резултат на инфекциозно заболяване. Главоболие, неразположение и треска с неясна етиология често предхождат началните фази на заболяването.
Последващите симптоми са подобни на тези при множествена склероза и могат да включват деменция, афазия, гърчове, нарушено внимание, тремор, нарушено равновесие, инконтиненция, мускулна слабост, главоболие, повръщане и увреждане на зрението и говора.
Други симптоми включват слабост от едната страна на тялото, мускулна скованост, проблеми със слуха и инконтиненция на функцията на червата и пикочния мехур.
Други мозъчно-стволови или церебеларни дефицити, които се срещат, включват световъртеж, офталмоплегия, нистагъм, лицева парализа, дизартрия или дисфагия.
Аномалиите на периферните черепни нерви, които понякога се срещат, включват оптичен неврит и оптична атрофия.
Кортикалната слепота е често срещана и могат да се открият различни отклонения, особено хемианопсия. Може да се открие хемипареза или кортикален сензорен дефицит.
Нерядко се срещат раздразнителност, промени в личността, объркване, дезориентация и поведенчески смущения. Пациентите може да изглеждат психотични.
Недохранването и кахексията обикновено се съобщават в средните или късните хронични стадии на заболяването, особено в случаите, когато при пациентите се развиват значителни нарушения на неврологичната функция, като например хронично вегетативно състояние.
Диагноза
Лабораторни изследвания
При рутинни лабораторни изследвания не се откриват специфични характеристики на това заболяване.
Резултатите от изследванията на серумните дълговерижни мастни киселини и изследванията на надбъбречната функция трябва да бъдат доказано нормални. Ако на отклонения в резултатите от тези изследвания персистират, трябва да се постави алтернативна диагноза адренолевкодистрофия.
В случаи, за които се подозира, че са примери за болест на Schilder, трябва да се направят електроенцефалография (ЕЕГ) и анализ на церебро-спиналан течност (ликвор).
Отклоненията в ЕЕГ, като периодични латерални епилептиформени разряди, предполагат алтернативна диагноза субакутен склерозиращ паненцефалит или прогресиращ рубеолен паненцефалит. Тези диагнози се подкрепят от установяване на отклонения в ликвора. Диагнозата на тези състояния се потвърждава чрез демонстриране на повишени титри за рубеола вирус в ликвора.
Трябва да се направят изследвания с цел установяване на възможна инфекциозна причина при остри или фулминантни случаи. Това усилие трябва да включва вирусни култури от ликвор, назален или орофарингеален секрет и ректално изследване.
Образни изследвания
Чрез ядрено-магнитен резонанс (ЯМР) се установяват 1 или 2 големи лезии в бялото вещество на мозъка.
Не трябва да се наблюдават допълнителни лезии при визуализиране на главен или гръбначен мозък, тъй като тази находка може да предполага наличието на множествена склероза, остър дисеминиран енцефаломиелит или някаква друга алтернативна диагноза.
Диагнозата никога не може да се основава само на образни характеристики. Находките от ЯМР, предполагащи наличието на болест на Шилдер, могат да бъдат най-често открити при пациенти с множествена склероза.
В такива случаи, в допълнение към големите лезии, по-малки лезии, подобни на множествена склероза, могат да бъдат открити при внимателно изследване, което предполага диагнозата на това, което Poser нарича преходна склероза, а не дифузна склероза.
Диагностични процедури
Може да са необходими мозъчни биопсични проби, за да се изключи инфекция, както и тумори, васкулитни или други възпалителни процеси (например първичен васкулит на ЦНС, саркоидоза, хистиоцитна лимфангиоматоза). Не може да се разчита на резултатите от мозъчна биопсия, за да се разграничи множествената склероза от острия дисеминиран енцефаломиелит.
Във всички случаи трябва да се извърши лумбална пункция, особено за да се изследват титри на антитела срещу морбили, за да се изключи алтернативната диагноза субакутен склерозиращ паненцефалит.
Лечение
Основният подход към лечението е да се установи дали състоянието реагира на прилагането на високи дози интравенозни кортикостероиди.
Практиката в такива случаи понастоящем включва приложение на метилпреднизолон за всяка от 3-5 последователни сутрини, последвано от намаляване на пероралните кортикостероиди. Това лечение може да бъде противопоказано поради възможното наличие на бактериални инфекции или неизключената възможност за херпесен енцефалит.
Прилагането на кортикостероиди трябва да бъде придружено от прилагане на антиациди или Н2-рецепторни блокери, за да се намали рискът от образуване на язви на Cushing. Пациентите трябва да бъдат наблюдавани за потенциални неблагоприятни ефекти като свръхчувствителност, стомашно-чревно кървене, хипертония, хипергликемия, хипокалиемия или опортюнистична инфекция.
Много от тези случаи изглежда реагират благоприятно на интравенозната стероидна терапия, въпреки че техният последващ курс вероятно ще бъде продължаващо развитие на пристъпи на множествена склероза или прогресивна форма на множествена склероза. В такива случаи трябва да се обмисли лечение с имуномодулиращи средства - бета-интерферон или имуносупресивна терапия.
В допълнение към лечението с кортикостероиди, терапията включва още подпомагане на дишането, циркулацията, храненето и внимание към поддържането на нормален метаболитен профили по време на острата фаза на заболяването.
Диетичен режим
Не е показана специфична диета за пациенти с болест на Schilder.
Активност
Няма конкретни ограничения за двигателната дейност. Пациенти, за които се смята, че има преходна склероза и следователно има вероятност да имат множествена склероза, могат да получат обостряне на симптомите на заболяване поради излагане на топлина, лошо хранене или преумора.
Усложнения
Усложненията включват церебрална херния, прогресиране на заболяването до смърт, развитие на пневмония, сепсис, белодробна емболизация, увреждане на кожата и язви и различни усложнения, дължащи се на прилагане на кортикостероиди.
Прогноза
Прогнозата на това заболяване е много променлива и може да има три различни курса: монофазен, без затихване; затихващ и прогресивен, с увеличаване на дефицита.
При тези индивиди, за които е докладвано, че имат болест на Schilder, средната продължителност на преживяемост е 6,2 години след началото (диапазон от 3 дни до 45 години). Продължителността на заболяването е по-малко от 1 година в 40% от случаите, но преживяемост от най-малко 10 години е наблюдавана в повече от 23%.
Тези случаи по всяка вероятност са примери за често агресивна форма на множествена склероза, поради което преживяемостта на тези индивиди обикновено е по-кратка от тази при повечето индивиди с множествена склероза.
В случаите в предпубертетна възраст с добър отговор към интравенозни кортикостероиди и особено тези, при които е установено, че имат по-малки лезии на места, типични за остър дисеминиран енцефаломиелит, може да е налице по-добра прогноза при някои случаи.
Симптоми и признаци при Дифузна склероза МКБ G37.0
ВсичкиЛечение на Дифузна склероза МКБ G37.0
Продукти свързани със ЗАБОЛЯВАНЕТО

Коментари към Дифузна склероза МКБ G37.0