Други факоматози, некласифицирани другаде МКБ Q85.8
Към групата заболявания "Други факоматози, некласифицирани другаде" се включват:
- Синдром на Peutz–Jeghers
- Синдром на Sturge–Weber
- Синдром на von Hippel–Lindau
 Синдромът на Peutz–Jeghers е рядко срещано заболяване, което се характеризира с кожно-лигавична хиперпигментация и интестинална полипоза. Израз е на вродена невроектодермална дисплазия. Женският пол е по-силно засегнат, като проявите се наблюдават от ранна възраст.
Синдромът на Peutz–Jeghers е рядко срещано заболяване, което се характеризира с кожно-лигавична хиперпигментация и интестинална полипоза. Израз е на вродена невроектодермална дисплазия. Женският пол е по-силно засегнат, като проявите се наблюдават от ранна възраст.
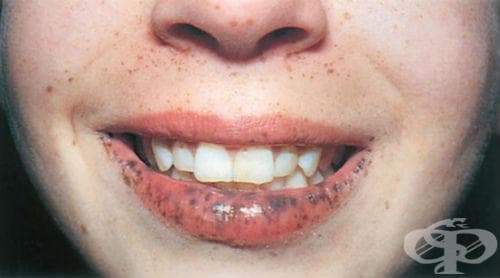
Първите симптоми на синдрома на Peutz–Jeghers са появата на множество тъмни петна по лицето и тялото, вариращи от жълто-кафяви до черни на цвят, локализирани основно периорално и периназално. Впоследствие се разпространяват по кожата на крайниците, главно по дланите и пръстите на ръцете. Петната са с овална форма и различна големина. Някои от тях изпъкват над повърхността на кожата, не избледняват при натиск.
Успоредно с пигментациите се наблюдава и полипоза на лигавицата на гастроинтестиналния тракт. Размерите на полипите варират – от едва доловими до големината на лешник, единични или множествени. Хистологично представляват хамартоми. Интестиналната полипоза може да има безсимптомно протичане, но в по-големия брой случаи са налице диспептични смущения – липса на апетит, гадене, тежест след нахранване, болки в корема, кръвоизливи.
Животозастрашаващите усложнения включват интестинална обструкция и увеличен риск от гастроинтестинални тумори (при 1/3 от болните полипите се израждат злокачествено). Съмнения за развитие на синдрома на Peutz–Jeghers пораждат наличието на тъмни петна по лицето, тялото и лигавиците. Диагнозата се потвърждава с гастроскопия или колоноскопия и установяването на чревна полипоза.
Поради съществуващия онкологичен риск е необходимо болните да бъдат внимателно и редовно проследявани.
 Синдромът на Sturge–Weber представлява енцефало-тригеминална ангиоматоза. Той е рядко наследствено заболяване, с честота 1:50 000. Дължи се на абнормно развитие на кръвоносни съдове в ембрионалния период. Съчетава кожна лицева и лептоменингеална хемангиоматоза. При малък процент от болните може да липсва кожен ангиом. Засегнатите съдове са най-често венозни.
Синдромът на Sturge–Weber представлява енцефало-тригеминална ангиоматоза. Той е рядко наследствено заболяване, с честота 1:50 000. Дължи се на абнормно развитие на кръвоносни съдове в ембрионалния период. Съчетава кожна лицева и лептоменингеална хемангиоматоза. При малък процент от болните може да липсва кожен ангиом. Засегнатите съдове са най-често венозни.
Кожните ангиоми обикновено се откриват веднага след раждането. Типичен е naevus flammeus в кожната зона на ramus ophthalmicus на n.trigeminus едностранно и рядко двустранно.
Състоянието протича с вродена глаукома. Често може да се открият локализирани аномалии в развитието на кръвоснабдяващия апарат на ретината.
През първата година започват фокални или вторично-генерализирани терапевтично резистентни епилептични припадъци. Други симптоми на заболяването са хемихипертрофия на езика, хемипареза, главоболие, зрителни нарушения и др. Възможни са прояви на умствено изоставане и мозъчни кръвоизливи.
Лечението е симптоматично с антиепилептични медикаменти. При тежки, застрашаващи живота епилептични припадъци, се извършва хирургична резекция на засегнатия лоб. Прогнозата е неблагоприятна.
 Синдромът на von Hippel–Lindau е рядко генетично мултисистемно неопластично заболяване с честота 1:40 000. Дължи се на двойна мутация на туморсупресорния ген VHL, разположен на късото рамо на трета хромозома. Характеризира се с ретинални и мозъчни хемангиобластоми, локализирани главно в малкия мозък, гръбначния мозък и мозъчния ствол, в съчетание с множествени кисти в различни органи, бъбречен карцином и феохромоцитом.
Синдромът на von Hippel–Lindau е рядко генетично мултисистемно неопластично заболяване с честота 1:40 000. Дължи се на двойна мутация на туморсупресорния ген VHL, разположен на късото рамо на трета хромозома. Характеризира се с ретинални и мозъчни хемангиобластоми, локализирани главно в малкия мозък, гръбначния мозък и мозъчния ствол, в съчетание с множествени кисти в различни органи, бъбречен карцином и феохромоцитом.
Честотата на различните тумори в състава на синдрома варира в широки граници в различните фамилии. Средната продължителност на живота е под 50 г.
Основните причини за летален изход при синдрома на von Hippel-Lindau, спадащ към групата на факоматози, некласифицирани другаде, са усложненията в резултат на бъбречноклетъчния карцином и хемангиобластомите на главния мозък.
Заболяването се изявява най-често между 20-30 годишна възраст. Манифестира се с развитие на хемангиоми в периферната част на ретината. Те се описват като окръглена червена маса и са свързани с разширяване и нагъване на артериален съд в ретината. Без лечение, а понякога и на фона на лечение, се наблюдава нарастване на ретинните хемангиобластоми с развитието на ексудативно отлепване на ретината и вторична глаукома.
Лечението в ранните стадии е добро. Прилага се лазерна коагулация или фотокоагулация на хемангиомите. При големи тумори ефективен метод е криотерапията. Оперативно лечение (витректомия) се прилага в случаите на отлепване на ретината или кръвоизлив в стъкловидното тяло. Прогнозата зависи от локализацията на туморите и настъпващите усложнения. Оставени без терапия, пациентите могат да загубят зрението си и да получат постоянни мозъчни увреждания. Своевременно поставената диагноза и навреме започнатото лечение значително подобряват прогнозата на заболяването.
Симптоми и признаци при Други факоматози, некласифицирани другаде МКБ Q85.8
- Повръщане
- Главоболие
- Ментална ретардация
- Безсимптомно протичане на заболявания
- Гърчове
- Затруднено дишане
Коментари към Други факоматози, некласифицирани другаде МКБ Q85.8