Факоматози, некласифицирани другаде МКБ Q85
Факоматозите или т.нар. неврокутанни синдроми са група наследствени заболявания, свързани с разстройство в развитието на ЦНС. Характеризират се с малформации и тумори в множество органи и системи – кожа, очи, централна и вегетативна нервна система.
Към групата заболявания "Факоматози, некласифицирани другаде" спадат:
- неврофиброматоза (болест на von Recklinghausen),
- туберозна склероза (болест на Bourneville, епилоя),
- цереброретинна ангиоматоза (синдром на von Hippel–Lindau),
- енцефалотригеминална невроангиоматоза (синдром на Sturge–Weber),
- синдром на Peutz–Jeghers и
- неуточнена факоматоза – хамартоза.
Всички тези заболявания показват фамилно разпространение и автозомно-доминантен начин на унаследяване с разнообразна експресивност.
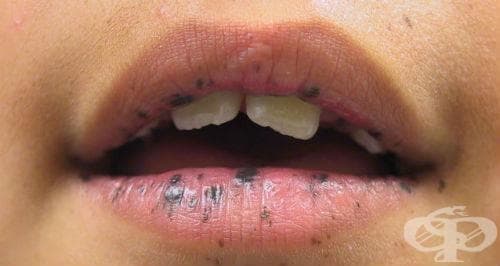
Синдромът на Peutz–Jeghers е рядко срещано заболяване, характеризиращо се с кожно-лигавична хиперпигментация и интестинална полипоза. Манифестира се с появяването на множество тъмни на цвят петна по лицето и тялото, вариращи от жълто-кафяви до черни, локализирани основно периорално и периназално. Интестиналната полипоза протича с диспептични смущения като липса на апетит, гадене, тежест след нахранване, болки в корема, кръвоизливи. Животозастрашаващите усложнения включват интестинална обструкция и увеличен риск от гастроинтестинални тумори.
Синдромът на Sturge–Weber представлява енцефало-тригеминална ангиоматоза. Той е рядко наследствено заболяване и съчетава кожна лицева и лептоменингеална хемангиоматоза. Типичен е naevus flammeus в кожната зона на ramus ophthalmicus на n.trigeminus едностранно и рядко двустранно. Друга характерна проява са фокалните или вторично-генерализираните терапевтично резистентни епилептични припадъци. Лечението е симптоматично с антиепилептични медикаменти. Прогнозата е неблагоприятна.
Синдромът на von Hippel–Lindau е рядко генетично мултисистемно неопластично заболяване. Характеризира се с ретинални и мозъчни хемангиобластоми, локализирани главно в малкия мозък, гръбначния мозък и мозъчния ствол, в съчетание с множествени кисти в различни органи, бъбречен карцином и феохромоцитом. Основните причини за летален изход са усложненията в резултат на бъбречноклетъчния карцином и хемангиобластомите на главния мозък. Лечението в ранните стадии е добро.
Неврофиброматозата или т.нар. болест на von Recklinghausen, отнасяща се към групата заболявания "Факоматози, некласифицирани другаде", бива две отделни, генетично и клинично различни форми – тип 1 и тип 2.
Неврофиброматоза тип 1 се среща по-често. Характеризира се с пигментации по кожата, тумори в централната и периферната нервна система, мозъчно-съдови промени и увреждане на вътрешните органи. Унаследява се автозомно-доминантно. Характеризира се с широка вариабилност на клиниката. Кожните промени са характерни. Налице са множество плоски светлокафяви петна тип „мляко с кафе“ (cafe-au-lait) с различна големина, които са налице от раждането и се увеличават след пубертета. Наблюдават се и хипопигментации, кожни тумори и подкожни неврофиброми, меланоцитни хамартоми в ириса (нодули на Lisch) и глаукома. Скелетни аномалии се установяват при около 1/3 от болните. Те включват черепни малформации, малформации на гръбначния стълб, сколиоза, кифоза. Диагнозата се поставя въз основа на клиничната картина и установените патологични промени в ЦНС чрез прилагане на КТ или ЯМР и разкриване на генния дефект.
Неврофиброматоза тип 2 е десет пъти по-рядка от тип 1. Заболяването най-често се изявява с тумори на централната нервна система. Около и след пубертета обикновено се установяват двустранни шваноми на вестибуларния нерв. Характерни симптоми са постепенна загуба на слух, шум в ушите, нарушен баланс. Не съществува специфично лечение.
Видове Факоматози, некласифицирани другаде МКБ Q85
Симптоми и признаци при Факоматози, некласифицирани другаде МКБ Q85
- Когнитивни увреждания
- Голяма глава
- Симптоми на кожата
- Кожни папули
- Подкожни подутини
- Аномалии на пикочо-половата система
Коментари към Факоматози, некласифицирани другаде МКБ Q85