Вродена ихтиоза, неуточнена МКБ Q80.9
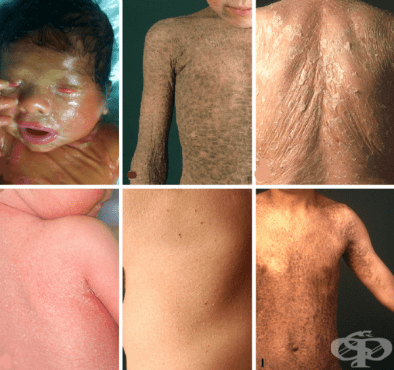
Това са група редки клинични форми на ихтиоза (Ichthyosis), при които ихтиотичните кожни промени са свързани с метаболитни и / или функционални и структурни аномалии.
Тук се отнасят:
- еритрокератодермия вариабилис (erythrokeratodermia variabilis);
- кератит - ихтиоза и глухота (KID) синдром (keratitis-ichthyosis and deafness syndrome);
- синдром C.H.I.L.D (CH - Congenital Hemidysplasia, I - Ichthyosiform Erythroderma, LD - limb defects).
Синдромите, включени в групата на вродена ихтиоза, неуточнена, са наследствени заболявания с различен начин на унаследяване - автозомно-доминантен, автозомно-рецесивен или полово обусловен (с Х - хромозомата).
Еритрокератодермия вариабилис (erythrokeratodermia variabilis)
Еритрокератодермия вариабилис е автозомно-доминантно наследствено заболяване, пациентът наследява мутацията от един засегнат родител. Други случаи са резултат от новонастъпили, спонтанни генни мутации и се наблюдават при хора, които нямат история на еритрокератодермия в семейството.
Състоянието се характеризира с хиперкератинизация на роговия слой на епидермиса, в съчетание с невъзпалителна еритема (раздразнена кожа).
Заболяването е рядко срещано и точното му разпределение не е известно, засяга еднакво и двата пола. При 90% от пациентите състоянието се развива до първата година, а при 50% е видимо веднага след раждането.
Еритрокератодермия вариабилис е причинена от мутации в гените GJB3 или GJB4. Тези гени кодират протеини, наречени конексин 31 и конексин 30.3. Те са част от семейство на протеини, които образуват канали по повърхността на клетките. Тези канали служат за регулиране на потока от хранителни вещества, заредени атоми (йони) и други сигнални молекули от една клетка към друга. Връзките, образувани с конексин 31 и конексин 30.3, се намират в най-външния слой на кожата. Анормалните протеини се натрупат в клетъчна структура, наречена ендоплазмен ретикулум, което води до индуциране на оксидативен стрес. Той довежда до клетъчна увреда и преждевременна смърт на корнеоцитите. Клетъчната смърт води до възпаление на кожата, което е в основата на развитието на еритематозните области.
Два основни симптома характеризират еритрокератодермия вариабилис:
- хиперкератоза;
- невъзпалителна еритема.
 Хиперкератозата е причина за образуването на сквами (люспи), те биват червеникаво - кафяви на цвят и могат да бъдат както дифузно разпространени по цялото тяло на пациента, така и да се появят само на малка площ. Люспите могат да варират по размер и форма, както и да се увеличават по размер с течение на времето. Областите на хиперкератоза (удебелена кожа) обикновено са симетрични - появяват се на едни и същи места от дясната и от лявата страна на тялото.
Хиперкератозата е причина за образуването на сквами (люспи), те биват червеникаво - кафяви на цвят и могат да бъдат както дифузно разпространени по цялото тяло на пациента, така и да се появят само на малка площ. Люспите могат да варират по размер и форма, както и да се увеличават по размер с течение на времето. Областите на хиперкератоза (удебелена кожа) обикновено са симетрични - появяват се на едни и същи места от дясната и от лявата страна на тялото.
Вторият характерен симптом на еритрокератодермия е наличието на еритематозни (раздразнени и зачервени) области. Обикновено са преходни и се различават по размер, форма и местоположение - нямат характерна локализация. Зачервяването може да се предизвика от внезапни промени в температурата, стрес, травма или дразнене на областта. Обикновено избледнява в рамките на часове до дни, не се съпровожда от усещане за болка. Трябва да се избягват излагане на студ, драстични температурни промени и механично дразнене на кожата.
Диференциалната диагноза се прави с други форми на ихтиоза, като епидермолитична ихтиоза, както и с ихтиозата на Кърт и Макин и KID синдром.
Диагнозата се поставя след провеждането на генетичен тест, анализиращ целият кодиращ район на гените GJB3 и GJB4.
В сравнение с останалите ретиноидни средства, ацитретинът (acitretin) е първо средство на избор при лечението на еритрокератодермия вариабилис.
Кератит-ихтиоза и глухота (КИГ) синдром
(keratitis-ichthyosis and deafness syndrome, (KID))
Синдромът на кератит, ихтиоза и глухота се установява веднага след раждането, протича със засягане на кожата, увреждане или двустранна загуба на слуха. Синдромът представлява автозомно-доминантно наследствено заболяване, причинено от мутации в конексин 26 - протеин, необходим за междуклетъчната комуникация.
Във вътрешното ухо канали, изградени от конексин 26, се намират в охлювна структура, наречена кохлея. Тези канали могат да помогнат за поддържането на необходимото ниво на калиеви йони, необходими за преобразуването на звуковите вълни в електрически импулси. Това превръщане е от съществено значение за нормалният слух.
В допълнение към клиничната картина, освен вродена двустранна загуба на слуха, кератит и еритрокератодермия, засегнатите индивиди също страдат от хронични бактериални и гъбични инфекции.
Възможно е развитието на кератит (възпаление на роговицата).
Кератитът може да причини:
- болка;
- чувствителност към светлина
- увеличаване на растежа на кръвоносни съдове
- прогресивна загуба на зрението;
- слепота.
 Пациентите страдат от палмоплантарна кератодермия (твърда кожа върху дланите на ръцете и стъпалата). В допълнение се наблюдават зачервени (еритематозни) петна по повърхността на кожата, които са сухи и люспести. Сквамите (люспите) могат да се появят навсякъде по тялото, но най-често се засягат областта около гърлото, слабините и подмишниците. Сквамозните участъци образуват цепнатини в епидермиса, които могат да бъдат населени от микроорганизми и да се развие бактериална инфекция. В тежки случаи тези инфекции могат да бъдат животозастрашаващи, особено в ранна детска възраст.
Пациентите страдат от палмоплантарна кератодермия (твърда кожа върху дланите на ръцете и стъпалата). В допълнение се наблюдават зачервени (еритематозни) петна по повърхността на кожата, които са сухи и люспести. Сквамите (люспите) могат да се появят навсякъде по тялото, но най-често се засягат областта около гърлото, слабините и подмишниците. Сквамозните участъци образуват цепнатини в епидермиса, които могат да бъдат населени от микроорганизми и да се развие бактериална инфекция. В тежки случаи тези инфекции могат да бъдат животозастрашаващи, особено в ранна детска възраст.
Други признаци на КИГ синдрома могат бъдат:
- области на плешивост (алопеция), които често засягат веждите или миглите;
- повишен риск от развитие на сквамозноклетъчен карцином на кожата (това се развива при около 12% от засегнатите индивиди);
- намалено изпотяване.
Дебелият рогов слой на кожата е причина и за намаляване на функцията на потните жлези, степента варира при пациентите, но често се развива хипохидроза (намалено отделяне на пот от организма). Което довежда до висок риск от прегряване на организма.
Диференциалната диагноза се прави с други клинични форми на ихтиоза (Ichthyosis).
Диагнозата се поставя след провеждане на генетичен тест, изследващ кодиращият район на гена за конексин 26, а именно дългото рамо на 13 хромозома.
КИГ синдрома изисква мултидисциплинарен подход чрез дерматологични (кожни), офталмологични (очни) и отоларингологични (УНГ) грижи.
Лосионите, съдържащи алфа-хидрокси киселини, могат да бъдат ефективно лечение на лющещата се кожата. Холестеролът или смесите, съдържащи серамид, също подобряват състоянието.
Оралните антибиотици и противогъбичните лекарства са необходими, при развитието на инфекции.
Необходимо е внимателно мониториране на кожата и лигавицата на устната кухина, поради риск от развитието на сквамозноклетъчен карцином.
Синдром C.H.I.L.D (C.H.I.L.D syndrome)
Синдромът е рядко срещано Х-свързано (с пола) наследствено заболяване, характеризиращо се с вродени дефекти в няколко системи - кожата, опорно-двигателния апарат и централната нервна система.
Синдромът C.H.I.L.D се появява спорадично, което означава, че само един член на семейството е засегнат. Рядко, състоянието може да се развие в няколко поколения и да се предава от майка на дъщеря. Среща се почти изключително при женския пол, защото фетусите (плода) от мъжки пол умират преди раждането.
Мутации в NSDHL гена причиняват C.H.I.L.D. синдром. Този ген кодира инструкции за синтеза на ензим, който участва в производството на холестерол. Мутациите, които са в основата на синдрома, елиминират активността на NSDHL ензима, което нарушава нормалното производство на холестерол в клетките.
Въпреки че високите нива на холестерола са известен рисков фактор за сърдечно-съдови заболявания, организмът се нуждае от така нареченият "добър" холестерол, за да се развива и да функционира нормално. Холестеролът е важен компонент при изграждането на клетъчните мембрани и на миелина.
Пациентите с левостранно засягане обикновено имат по-тежки аномалии на вътрешните органи, особено по отношение на сърцето. Високата смъртност най-често се дължи на сърдечносъдови малформации. Въпреки това, дефекти в развитието на централната нервна система, скелета, бъбреците, белите дробове и други могат да увеличат риска при тези пациенти.
Симптомите на това заболяване обикновено са ограничени по дясната или лява страна на тялото - хемидисплазия ("хемиа" означава "половина", "дисплазия"- анормален растеж), лицето обикновено не се засяга.
 Кожните аномалии се наблюдават при раждането и продължават през целия живот. Кожата е зачервена и възпалена (еритродермия).
Кожните аномалии се наблюдават при раждането и продължават през целия живот. Кожата е зачервена и възпалена (еритродермия).
Еритематозните участъци са покрити от дебели сквами (люспи), което свидетелства за ихтиозно състояние.
Аномалиите на крайниците се проявяват от страната на тялото, където се манифестират и аномалиите на кожата. Тези дефекти могат да варират от недостатъчно развитие на пръстите на ръцете и краката до пълното отсъствие на крайник.
Възможно е също така да възникнат аномалии от страна на централната нервна система (поради липса на миелинизация на нервните влакна), кръвоносните съдове, бъбреците, щитовидната жлеза, белите дробове и надбъбречните жлези, както и на репродуктивната и пикочната система. Тези аномалии са резултат от недостатъчно развитие на засегнатата страна на тялото.
Диференциалната диагноза се прави с останалите клинични форми на ихтиоза (след физикален преглед при специалист) поради характерния фенотип (външен вид) на пациентите диагнозата се поставя бързо.
За окончателното потвърждение на диагнозата е необходимо провеждането на генетичен тест, изследващ кодиращият район на NSDHL гена и тест за делеции и дупликации в този район.
Дерматологичните симптоми се лекуват чрез прилагане на омекотяващи мехлеми, за предпочитане са вазелинът и всички лосиони на основата на салицилова киселина.
Ретиноидите като третиноин (tretinoin) и еретинат (eretinat), могат да са ефективни, но в някои случаи могат да причинят токсични ефекти върху костите.
Лечението на останалите симптоми е поддържащо и е необходима намеса от специалисти от съответните медицински специалности.
Синдром на Нетертън (Netherton syndrome)
Синдромът представлява автозомно-рецесивно заболяване, чиято изява засяга нормалния строеж на кожата, космените фоликули и имунната система. Новородените със синдром на Netherton имат кожа, която е червена и люспеста (ихтиозоформена еритродермия).
Синдромът се среща приблизително при 1 на всеки 200 000 новородени.
Синдромът на Netherton се причинява от мутации в ген SPINK5. Този ген кодира инструкции за създаване на протеин, наречен LEKT1. Той отговаря за синтезата на протеини, които контролират активността на ензимите, наречени серин пептидази. LEKT1 се намира в кожата и в тимуса (жлеза, разположена зад гръдната кост, която играе важна роля в имунната система, като произвежда бели кръвни клетки, наречени лимфоцити). LEKT1 контролира активността на някои серинови пептидази във външния слой на кожата (епидермиса), особено твърдата външна повърхност, позната като stratum corneum, която осигурява здрава бариера между тялото и неговата среда. Също участва в нормалния растеж на косата, развитието на лимфоцити в тимуса и контрола на пептидазите, които задействат функцията на имунната система.
Кожата на новородените със синдром на Netherton е червена и люспеста (ихтиозиформена еритродермия). Някои засегнати бебета се раждат с обвивка, покриваща тялото им, наречена колоидална мембрана. Тази мембрана обикновено се отделя през първите няколко седмици от живота.
Поради това, че новородените с това разстройство нямат защита, осигурена от нормалната кожа, те са изложени на риск от дехидратация и развитие на инфекции, които могат да бъдат животозастрашаващи.
След ранна детска възраст тежестта на кожните аномалии варира. Кожата може да продължи да е зачервена и люспеста, особено през първите няколко години от живота.
Сърбежът е често срещан проблем за засегнатите лица. Надраскването може да доведе до чести инфекции поради нарушаване на роговия слой на епидермиса.
Дебелият рогов слой на кожата е причина и за намаляване на функцията на потните жлези, поради това е възможно засегнатите индивиди да имат затруднения при регулиране на телесната си температура.
 Пациентите със синдром на Нетертън имат коса, която е крехка и се разпада лесно. Тази характеристика е известна като коса от бамбук, трихорексия нодоза (trichorrhexis nodosa) или трихорексис инвагината (trichorrhexis invaginata). Нарича се така поради характерния вид на космите при наблюдение под микроскоп. Освен космите на скалпа, миглите и веждите могат да бъдат засегнати.
Пациентите със синдром на Нетертън имат коса, която е крехка и се разпада лесно. Тази характеристика е известна като коса от бамбук, трихорексия нодоза (trichorrhexis nodosa) или трихорексис инвагината (trichorrhexis invaginata). Нарича се така поради характерния вид на космите при наблюдение под микроскоп. Освен космите на скалпа, миглите и веждите могат да бъдат засегнати.
От страна на имунната система често срещани са хранителните алергии, сенната хрема или периодични прояви на възпаление на кожата (екзема).
Изследването на косми от коса, под микроскоп, показва трихорексия инвагината (trichorrhexis invaginata) - наблюдават се дефекти в строежа на космите. За потвърждаване на диагнозата може да се извърши и кожна биопсия.
Няма специфично лечение за синдром на Netherton. Целите на лечението са да се контролират симптомите и да се предотвратят кожни инфекции или други усложнения.
Трябва да се прилагат емолиенти и кератолитици (например кремове, съдържащи урея, млечна киселина или салицилова киселина), за да се поддържа влажна и хидратирана кожа.
Антибиотиците могат да се използват при наличие на кожни инфекции.
Локалните стероиди могат да бъдат полезни при по-големи деца с екзема.
Библиография
http://emedicine.medscape.com/article/1110820-overview#showal
https://medlineplus.gov/geneticcounseling.html
https://www.ncbi.nlm.nih.gov/gtr/conditions/C0265961/
https://ghr.nlm.nih.gov/condition/erythrokeratodermia-variabilis-et-progressiva#diagnosis
https://www.ncbi.nlm.nih.gov/pubmed/2150049
https://www.ncbi.nlm.nih.gov/pubmed/25546246
https://ghr.nlm.nih.gov/condition/keratitis-ichthyosis-deafness-syndrome
http://www.firstskinfoundation.org/types-of-ichthyosis/keratitis-ichthyosis-deafness-kid
https://ghr.nlm.nih.gov/condition/congenital-hemidysplasia-with-ichthyosiform-erythroderma-and-limb-defects#inheritance
Коментари към Вродена ихтиоза, неуточнена МКБ Q80.9