Вродени пороци в развитието на опорно-двигателния апарат
Вродените пороци на опорно-двигателния апарат заемат важно място в патологията на детската, като се срещат в 13-14% от починалите в перинаталния период деца. Особеност при тях е фактът, че те почти винаги представляват компонент на множествени вродени аномалии и много рядко се срещат изолирано, в чист вид. Сред вродени пороци в развитието на опорно-двигателния апарат най-голям дял се пада на аномалиите на крайниците.
Вродените пороци на костната система биват:
- системни - ахондроплазия, ахондрогенеза, фиброзна дисплазия, артрогрипоза, остеогенезис имперфекта
- локални - амелия, фокомелия, хемимелия, монодактилия, брахидактилия, полидактилия, синдактилия, полифалангия, синостоза, сиреномиелия
Скелетните дисплазии са хетерогенна група от заболявания, характеризиращи се с присъщи аномалии в растежа и/или ремоделирането на хрущялите и костите. Тези дисплазии засягат черепа, гръбначния стълб и крайниците в различна степен. Те често причиняват непропорционално малък ръст (джудже). Ахондроплазията е най-честият вид непропорционален дарфизъм с къси крайници.
Терминът ахондроплазия, предполагащ отсъствие на хрущял, е използван за пръв път от Parrot през 1878 година. Ахондроплазията се причинява от мутация в рецептора на фибробластния растежен фактор 3 (FGFR3). При нормално развитие той има отрицателен регулаторен ефект върху растежа на костите. При ахондроплазия, мутиралата форма на рецептора е конститутивно активна и това води до силно съкратени кости. Ефектът е генетично доминантен, като едно мутантно копие на FGFR3 генът е достатъчно да причини ахондроплазия, докато две копия на мутантния ген са неизменно фатални (рецесивно смъртоносни) преди или малко след раждането (известно като летален алел). Хората с ахондроплазия могат да бъдат родени от родители, които нямат състояние поради спонтанна мутация.
Изследванията показват, че нови генни мутации за ахондроплазия са само наследени от бащата и възникват по време на сперматогенезата. Теоретично, оогенезата има някакъв регулаторен механизъм, който предотвратява предаването на мутациите при жените.
Основният дефект, открит при пациенти с ахондроплазия, е абнормна ендохондрална осификация. Периостеалната и интрамембранозната осификация е нормална. Тръбните кости са къси и широки, отразяващи нормалния периостален растеж. Илиачните гребени се апофизират (растежът на аполиаза) са нормални, което води до големи, квадратни илиачни крила. Растежът на трирадиатния хрущял (ендохондрален растеж) е ненормален, което води до хоризонтални ацетабуларни покриви. По този начин, моделите на дефекта помагат да се обяснят много от наблюдаваните клинични и рентгенографски характеристики на ахондроплазията.
Растежът на трирадиатния хрущял (ендохондрален растеж) е ненормален, което води до хоризонтални ацетабуларни покриви. По този начин, моделите на дефекта помагат да се обяснят много от наблюдаваните клинични и рентгенографски характеристики на ахондроплазията.
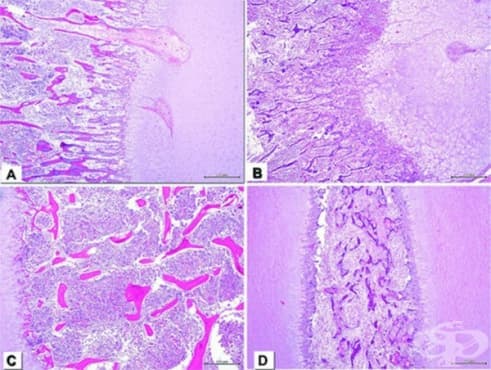
Биопсията от растежните пластини на илиума и проксималната фибула разкрива по същество нормална структура (фигура 1). Определянето на глюкозаминогликан е нормално. Делът на протеогликановите агрегати се увеличава във фибуларната глава. Дефектът е предимно количествен и се намира в пролиферативната зона на плаката за растеж.
Растежната плоча при ахондроплазията е силно изтъняла, а зоната на пролиферативния хрущял или липсва, или силно отслабва. Зоната на временната калцификация, ако присъства, претърпява ендохондрална осификация, но с много намалена скорост.
Остеогенезис имперфекта, известна също като крехка костна болест, е група от генетични нарушения, които засягат главно костите. Това води до кости, които лесно се разрушават. Тежестта може да е лека до тежка. Други симптоми могат да включват сини склери, чупливост на костите, хлабави стави, загуба на слуха, проблеми с дишането и проблеми със зъбите. Усложненията могат да включват дисекация на цервикалната артерия и дисекция на аортата.
При остеогенезис имперфекта, спадаща към вродени пороци в развитието на опорно-двигателния апарат се наблюдават патологични промени във всички тъкани, от които колаген тип 1 е важна съставка (например кости, лигаменти, дентин и склера). Основният дефект е едно от качественото или количествено намаляване на колаген тип 1. Мутациите в гените, кодиращи колаген тип 1, засягат кодирането на един от двата гена, което представлява приблизително 80% от случаите на заболяването.
Повечето случаи на заболяването, по-рано смятани за автозомно доминантно или автозомно рецесивно, сега са известни като възникващи от автозомни доминантни мутации. Наследствените мутации имат риск от повторна поява при последващи бременности от 50%, ако един от родителите е засегнат, докато новите мутации имат непредсказуем риск от повторна поява. Малък брой от случаите, за които се предполага, че са автозомни рецесивни, вече са доказани чрез молекулярен и свързващ анализ, който е вторичен за гонадалния мозайцизъм. Рискът от повтаряне на тези случаи също е непредсказуем.
При костите степента на хистологична промяна корелира добре с клиничната тежест на заболяването. Болестта засяга както ендохондралната, така и интрамембранозната осификация.
При остеогенезис имперфекта поради количествени дефекти на колаген тип 1 възниква лека форма на заболяването. При светлинна микроскопия е налице остеопоротична кост с дебели остеоидни шевове и намалена междуклетъчна матрица (фигура 2). Броят на остеокластите и остеоцитите е нормален. Костните трабекули са тънки и дезорганизирани. Ламеларната кост се вижда в диафизата и метафизата. При електронна микроскопия, остеобластите показват разширена ендоплазматичен ретикулум (вероятно порадинатрупване на непълни молекули на проколаген), а колагеновите влакна са с намален диаметър.
При остеогенезис имперфекта поради качествени дефекти на колаген тип 1 се наблюдава тежка форма на заболяването. Светлинната микроскопия разкрива хиперостеоцитоза и повишени съдови канали. Други открития са намалената дебелина на костната кост, липсата на нормално образуване на кортикална кост и дезорганизирането на плаката за растеж (фигура 3). Електронната микроскопия показва слабо запазени остеобласти и колагенни връзки с променлив диаметър, особено в по-смъртоносните форми на заболяването.
Епифизата и физата са склонни да бъдат широки и неправилни, с дезорганизация на пролиферативните и хипертрофичните зони и загуба на типичната колонна подредба. Изяснява се изтъняването на зоната на калциевия хрущял, заедно с недостиг на първична спонгиоза на метафизата и забавяне на вторичните центрове на осификация в епифизата.
Ширината на биопсичните сърцевини, ширината на кортекса и обемът на изследваните кости са намалени при всички видове остеогенезис имперфекта. Броят и дебелината на трабекулите са намалени. Пробите могат да показват доказателства за дефекти в моделирането на външната кост по отношение на размера и формата, производството на вторични трабекули чрез ендохондрална осификация и уплътняването на вторичните трабекули чрез ремоделиране. Следователно, остеогенезис имперфекта може да се разглежда като заболяване на остеобластите.
Количеството на костите се понижава, но качеството на костния материал вероятно е най-важно в патогенезата на заболяването.

Продукти свързани със СТАТИЯТА

КАЛЦИЙ 400 мг + ВИТАМИН D3 таблетки * 150 SANCT BERNHARD

ХАЯ ЛАБС ВИТАМИН D3 капсули 1000 IU * 100
ПРОМО
НАУ ФУДС КОЛАГЕН UC - II ТИП II капсули 40 мг * 60

РИБЕНО МАСЛО ОТ ЧЕРЕН ДРОБ НА ТРЕСКА С ОМЕГА 3 И ВИТАМИНИ A И D капсули 1000 мг * 120 HOLLAND & BARRETT

МАГНЕМАТРИКС таблетки * 30 ВИТАГОЛД

КАЛИВИТА ДЖОЙНТ ПРОТЕКС ФОРТЕ таблетки * 90
Безплатна доставка за България!Библиография
Color atlas of pathology, Section Congenital malformations
https://emedicine.medscape.com/article/1258401-overview
https://en.wikipedia.org/wiki/Achondroplasia
https://emedicine.medscape.com/article/1256726-overview#a3
https://en.wikipedia.org/wiki/Osteogenesis_imperfecta
https://emedicine.medscape.com/article/1256726-workup#c10
СТАТИЯТА е свързана към
- Обща патология
- Смущения в развитието на организма
- КП № 265 ФИЗИКАЛНА ТЕРАПИЯ И РЕХАБИЛИТАЦИЯ ПРИ БОЛЕСТИ НА ОПОРНО-ДВИГАТЕЛЕН АПАРАТ
- д-р Красимир Йорданов Великов
- д-р Христо Димитров Вълчев
- д-р Иван Веселинов Бончев
- д-р Любомир Спасов
- Костна тъкан
- д-р Димитър Любенов Димитров
- д-р Севдалина Лазарова Йоцова
- д-р Надежда Димитрова Колева-Манечкова
- д-р Иванка Борисова Томова
Коментари към Вродени пороци в развитието на опорно-двигателния апарат