Синдроми на вродени аномалии, свързани предимно с нисък ръст МКБ Q87.1
Ръстът е най-важния показател за физическо развитие, тъй като се определя предимно от ендогенните фактори. Като нисък ръст може да се дефинира ръст с 2 и повече стандартни отклонения, под средната норма за съответната възраст (SD).
Рубриката "Синдроми на вродени аномалии, свързани предимно с нисък ръст" включва:
- синдром на Aarskog
- синдром на Cockayne
- синдром на De Lange
- синдром на Dubowitz
- синдром на Noonan
- синдром на Prader-Willi
- синдром на Robinow-Silverman-Smith
- синдром на Russel-Silver
- синдром на Seckel
- синдром на Smith-Lemli-Opitz
Рубриката не включва синдром на Ellis-van Creveld, който се разглежда в:
Синдром на Аарског-Скот (САС) е един от синдромите на вродените аномалии, свързани предимно с нисък ръст. То е много рядко генетично заболяване, причинено от мутация в Х-хромозомата. Съществува също така автозомно-доминантен и автозомно-рецесивен тип на заболяването. Очакваната честота е 1:25 000. В световната литература са описани 40 случая. Засяга предимно мъже. Като при жените заболяването протича в сравнително по–лека форма. Условието за развитие на заболяването е наличието на мутации в ген, наречен "лицево-генитална дисплазия" (FGD1; Xp11.21).
Симптомите на заболяването включват:
- стърчащ напред пъп
- офанзива в слабините или скротума
- забавена полова зрялост
- забавено никнене на зъби
- напречна гънка в долната устна
- ниско разположени уши (с форма на чаша)
- страбизъм
- линията на косата е с "връх вдовица"
- лека до умерена умствена изостаналост
- леко до умерено изоставане на височина, което е трудно забележимо от 1 до 3 години
- слабо развита средна част на лицето
- скротума заобикаля пениса, като „шал“
- незавършен десцензус на тестисите
- къси пръсти на ръцете и краката с леки извивки по тях като „лебедова шия“
- единична гънка в дланта на ръката (маймунска бразда)
- малки, широки ръце и крака с къси пръсти и извит пети пръст
- малък нос с изпъкнали ноздри напред
- обърнати ноздри, максиларна хипоплазия
- леко прегъната горна част на ухото
- бразда над горната устна, мачка под долната устна
- очи с увиснали клепачи
- частична синдактилия (ремъци), и удължение - клинодактилия
- когнитивни и поведенчески разстройства, които често са ограничени до ранна детска възраст
- интелектуални нарушения, рядко са тежки
- проблеми в ученето и дефицит във вниманието
- хиперактивност
При раждането, размерите на тялото в повечето случаи са нормални, но по време на ранна детска възраст и детството се забавят. Може да е причина за забавен растеж преди пубертета. Задълбочаването на растежа в края юношеството води до умерено нисък ръст. Гениталните аномалии могат да включват крипторхизъм, макроорхизъм, по-рядко хипоспадия. При засягане на женския пол най–често се наблюдават "Връх вдовица" и хипертелоризъм. Пациентите могат да страдат от невропсихологични смущения.
Клиничната диагноза е подкрепена от данните от прегледа, от присъствието на най-характерните клинични признаци и молекулно-генетичен анализ на ген FGD1. Наличието на всички данни потвърждават диагнозата синдром на Аарског. В случаите, в които молекулярният анализ не потвърди диагнозата, е необходимо да се изключи подобен.
Пренатална диагноза и определяне на риска от заболяване е възможно, когато мутацията за съответната фамилна форма е известна(повечето мутации са специфични за всяко семейство). В генетична консултация тя изисква дълбоко проучване на семейната история на пациента.
Ефективното лечение на САС не съществува. За повечето пациенти прогнозата е благоприятна. Обикновено такива пациенти безопасно достигат до зряла възраст.
Кокейн синдромът (Cockayne; CS) е рядка форма на нанизъм. Това е наследствено заболяване, чиято диагноза зависи от наличието на три признака:
- забавяне на растежа, т.е. нисък ръст
- анормална чувствителност към светлина (фоточувствителност)
- старчески вид (progeria).
Генът, отговорен за CS-тип I е съпоставен с хромозома 5 и се нарича ERCC8. Генът за CS-тип II е съпоставен с хромозомен локус 10q11 и се нарича ERCC6. Мутации в ERCC6 се откриват при около 75% от случаите, докато мутации в ERCC8 причиняват около 25% от случаите. Характерно е АР унаследяване.
Основните характеристики на синдрома на Cockayne включват: спиране или забавяне на нормалния растеж (нанизъм), по време на късния начален стадий, изключителна чувствителност към светлина (фоточувствителност) и старчески вид (прогериа). Кожата изглежда набръчкана и възрастна, особено по лицето, ръцете и краката, поради загубата на подкожна мастна тъкан. Децата с това заболяване могат да се наранят по-лесно и имат по-интензивна пигментация на кожата.
Различните видове на заболяването се определят спрямо възрастта, в която е започнало заболяването.
- CS тип I, класическата форма, се характеризира с нормално новородено, чиито симптоми не могат да станат ясни преди първата година. Височина и тегло, както и други показатели за размер и растеж са много под персантила, за съответната възраст. Зрение, слух, и нервната система, функционираща (централна и периферна) се влошават с течение на времето. Развива се тежка инвалидност.
- Малка част от случаите на вроден CS тип II, се характеризират с очевидни нарушения в растежа при раждането заедно с малко или никакво засягане на неврологичното развитие след раждането. Сериозни нарушения в зрението (катаракта и други структурни аномалии на окото) обикновено присъстват при раждането. Рано се наблюдават скелетни аномалии.
- CS тип III е по-рядка и се характеризира с по-нормален растеж и умствено развитие през първите години, които се прекъсват от късната поява на типичните симптоми на CS.
- XP-CS е най-рядка форма и включва характеристики на двете заболявания. Зоните на хиперпигментации и началото на рак на кожата са типични за ксеродерма и нисък ръст, умствено изоставане и сексуална недоразвитост са в съответствие с CS.
Децата с Cockayne синдром имат необичайни физични характеристики: малка глава (микроцефалия), необичайно тънък нос, "кух" или хлътнал вид на очите, големи безформени уши, лошо затваряне на клепача, и / или ненормална проекция на горните и долните челюсти (прогнатизъм). Възможно е да има необичайно големи кариеси, поради необичайно развитие на зъбите. Засегнатите лица обикновено са с големи ръце и крака, и необичайно дълги ръце и крака. Ставите също могат да са необичайно големи и остават във фиксирано положение (флексия), гръбначния стълб може да бъде извит навън, когато се гледа отстрани (кифоза). Други характеристики на Cockayne синдром могат да включват намалено потене (хипохидроза), намалено сълзене на очите, и / или преждевременно побеляване на косата, ненормално син оттенък на кожата (цианоза) на ръцете и краката.
Неврологичните симптоми се проявяват с ритмични, трептящи движения (тремор), нестабилна походка (атаксия), и / или невъзможност да се координират движенията. Засегнатите деца могат да получат различна степен на умствена изостаналост, частична загуба на слуха, и / или прогресивна загуба на придобити преди интелектуални способности.
Пораженията върху очите включват прогресивно помътняване на лещата на очите (катаракта), загуба на зрение (атрофия на зрителния нерв), дегенерация на ретината, и / или ненормално натрупване на ретината оцветяване (пигментация).
Някои хора с Cockayne синдром може да имат необичайно високо кръвно налягане (хипертензия), увеличение на черния дроб (хепатомегалия), и / или преждевременно натрупване на мастни плаки върху стените на артериите около сърцето (атеросклеротична болест). Възрастни с това заболяване могат да бъдат сексуално недоразвити.
Лечението на Cockayne синдрома е симптоматично и поддържащо. Генетична консултация може да се препоръча на членовете на семейството.
Синдромът на Корнелия де Ланге (CdLS) е разстройство на развитието, което засяга много части на тялото. Може да е резултат от мутации в най-малко пет гени: NIPBL, SMC1A, HDAC8, RAD21, и SMC3. Мутации в гена NIPBL са били идентифицирани в повече от половината от всички хора с това заболяване. В около 30% от случаите, причината за синдрома на Корнелия де Ланге е неизвестен. Изследователите търсят допълнителни промени в пет известни гени, както и мутации в други гени, които могат да предизвикат това състояние.
Характеристиките на това заболяване, включващо се към рубриката "Синдроми на вродени аномалии, свързани предимно с нисък ръст", варират от сравнително леки до тежки. Синдромът на Корнелия де Ланге се характеризира с бавен растеж, преди и след раждането води до нисък ръст; умствено увреждане, което обикновено е умерено до тежко и аномалии на костите на ръцете, ръцете и пръстите. Повечето хора със синдром на Корнелия де Ланге също имат отличителни черти на лицето, включително извити вежди, дълги мигли, ниско поставени уши, малки и широко разположени зъби и малък, обърнат нагоре нос. Много от засегнатите лица имат проблеми с поведението, подобни на аутизъм, състояние на развитие, което засяга комуникацията и социалното взаимодействие.
Допълнителни признаци и симптоми на синдрома на Корнелия де Ланге може да включват прекалено окосмяване по тялото (хипертрихоза), необичайно малка глава (микроцефалия), загуба на слуха и проблеми с храносмилателния тракт. Някои хора с това заболяване се раждат с отвор в покрива на устата, който се нарича цепка на небцето. Гърчове, сърдечни дефекти и проблеми с очите също са били докладвани при хора с това заболяване.
Синдромът на Dubowitz е много рядка генетична аномалия в развитието. Това разстройство включва множество вродени (наследени) аномалии, но не само: изоставане в растежа / нисък ръст; необичайни, но характерните черти на лицето; малка глава (микроенцефалия); лека умствена изостаналост; и, в най-малко 50% от случаите, екзема. Множество органи и системи са засегнати и разстройството е непредсказуем и изключително променлива в изразяването му. Симптомите могат да бъдат открити, докато плодът е все още в матката (вътрематочно), както и веднага след раждането (неонатална).
Външен вид на лицето е от ключово значение за диагнозата, с характерна високо или наклонено чело; рядка коса; плоски, неразвити (хипопластични) кости над очите (надочничните хребети); увеличава разстоянието между очите (очна хипертелоризъм); увиснали клепачи (птоза); хипопластични странични вежди; много малка долна челюст (микрогнатия) и отдръпването на брадичката (ретрогнатия). Засегнатите деца често са хиперактивни, упорити и срамежливи.
Много деца със синдром на Dubowitz имат нормален интелект. Въпреки това, могат да се появят трудности с паметта и / или когнитивни увреждания. Засегнатите лица могат да имат леко забавяне, хиперактивност, и / или говор увреждане. Може също да се получи известно увреждане на глоба двигателното развитие и когнитивно мислене.
Диагнозата обикновено се базира на клинично решение, изготвено от генетик или лекар. Тя се основава на медицинската история на пациента, данни за растеж и особено на лицето вид характеристика.
Лечението на синдрома на Dubowitz е симптоматично и поддържащо. Генетичното консултиране може да бъде от полза за засегнатите индивиди и техните семейства.
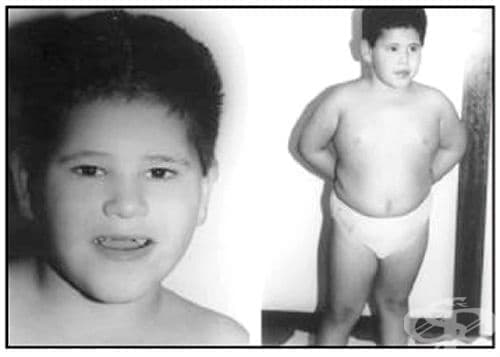
Синдромът на Нуунан (Noonan) е автозомно-доминантно заболяване, което се характеризира с нисък ръст, типичен лицев дисморфизъм и вродени сърдечни аномалии. В постнаталния период се установяват стигми като: широко чело, хипертелоризъм, епикант, антимонголоидни очни цепки, ниско разположени ушни миди, уплътнен хеликс, високо разположено небце, микрогнатия, къс врат с кожни гънки и ниско разположена инсерция на косата.
Контурите на лицето обикновено стават триъгълни с напредване на възрастта. У деца лицето може да изглежда миопатно, с проминиращи очни ябълки, уни- или билатерална птоза, изразени назолабиални гънки и тънка уста. В млада възраст очите са по-малко изпъкнали, а вратът не изглежда толкова къс. Обикновено, по-възрастните индивиди са с изразени назолабиални гънки, висока предна линия на косата, дебели отпуснати клепачи и набръчкана кожа. В по-късна възраст и особено при възрастни пациенти лицевите особености са минимални. Най-честата сърдечна аномалия е пулмоналната клапна стеноза с диспластични платна (50–62) Дължината и теглото обичайно са нормални при раждането.
Теглото може да бъде по-високо поради подкожните отоци. В този случай през първите няколко седмици се установява значителна редукция. Проблеми със сукането и неонаталното хранене възникват при 63% от болните, но обикновено се разрешават спонтанно през ранното детство. Пубертетът се забавя с около две години, а пубертетният растежен скок е намален или липсва. Костната възраст също изостава с около две години. Средният ръст при мъже е 162.5 см, а при жени – 152.7 см. И двете стойности са под 3-ти сентил за здравата популация.
Други характерни деформации включват:
- Гръден кош: пектус каринатус и пектус екскаватус, широк, мамилите са раздалечени.
- Полови смущения: крипторхизмът е много чест; Обикновено ЛХ и ФСХ са високи, спермалните нарушения. И при двата пола пубертетът е забавен, средната възраст на менархе е 14.6 години. При жените обаче не се наблюдава инфертилитет.
- Смущения в отделителната система: Малформации - най-чести са пиелоуретралните стенози с или без хидронефроза. Кръвонасяданията и кръвоизливите са чести. Изследванията на коагулацията показват удължено време на кървене, дефицит на фактори VIII, XI и XII, тромбоцитопения и дефекти в тромбоцитната функция.
- Нарушения в пигментацията.
- Очни нарушения - страбизъм, рефракционните аномалии и амблиопията
- Нарушенията на слуха - възпаление на средното ухо
- Коремни органи: Хепатоспленомегалия се среща често. Тя не е свързана с чернодробна или спленална дисфункция, нито със сърдечна недостатъчност.
- Нарушения във физическото развитие: децата с NS показват леко забавено моторно развитие, което отчасти се дължи на мускулната хипотония, типична в ранното детство. Средната възраст, на която децата сядат, е 10 месеца, ходят самостоятелно – на 21 месеца, а проговарят – на 31 месец.
- Менталната ретардация се характеризира със специфични визуално-конструкционни проблеми и вербални нарушения. Средният коефициент на интелигентност е 85. Основните поведенчески проблеми включват непохватност, хранителни разстройства, нервност, инатливост, раздразнителност и ехолалия. Срещат се също социални проблеми и нарушения във вниманието.
За правилната му диагноза е разработена помощна точкова система. По-голямата част от децата с NS ще пораснат и живеят нормално в света на възрастните.
Синдромът на Прадер-Уили (Prader Willi; PWS) е сложно генетично заболяване, което засяга много части на тялото. Причината се дължи на загубата на функция на гени в даден регион на хромозома 15. Хората обикновено наследяват едно копие на тази хромозома от всеки родител. Някои гени са включени (активни) само върху копието, което е наследено от бащата на лицето (бащино копие). Това специфично генно активиране се причинява от феномен, наречен геномно отпечатване. Повечето случаи на синдром на Прадер-Уили не се унаследяват, особено тези, причинени от делеция в бащина хромозома 15 или по майчина дизомия. Тези генетични промени се срещат като случайни събития по време на формирането на репродуктивните клетки (яйцеклетки и сперматозоиди) или в ранното развитие на плода. Засегнатите хора обикновено нямат никаква история на заболяването в семейството им.
Рядка, генетична промяна, отговорна за синдром на Прадер-Уили може да се наследява. Например, че е възможно за генетична промяна, която необичайно инактивира гени на бащина хромозома 15, за да се предават от едно поколение на друго.
В ранна детска възраст, това състояние се характеризира със слаб мускулен тонус (хипотония), трудности при хранене, слаб растеж и забавено развитие. В началото на детството, засегнатите индивиди имат ненаситен апетит, което води до хронично преяждане (хиперфагия) и затлъстяване. Някои хора със синдром на Prader-Willi, особено тези със затлъстяване развиват диабет тип 2 (най-честата форма на диабет).
Хората със синдром на Прадер-Вили, спадащ към синдроми на вродени аномалии, свързани предимно с нисък ръст, обикновено имат лека до умерена интелектуална недостатъчност и когнитивни увреждания. Поведенческите проблеми са общи, включително изблици темперамент, упоритост и натрапчиво поведение, като бране на кожата. Аномалии на съня също може да се появят. Допълнителни дисфункции на това състояние включват отличителни черти на лицето като тясно чело, бадемови очи и триъгълна уста; нисък ръст и малки ръце и крака. Някои хора със синдром на Прадер-Уили имат необичайно светла кожа и светла коса. И двата пола мъже и жени имат недоразвити гениталии. Пубертетът е забавен или непълен. Най-засегнатите лица не могат да имат деца (безплодни).
Синдромът на Robinow-Silverman-Smith (Robinow нанизъм) е изключително рядко генетично заболяване, характеризиращо се с къси крайници, нанизъм, аномалии в главата, лицето и външните полови органи, както и гръбначната сегментация. Съществуват две форми на заболяването - доминантна и рецесивна, от които първата е по-честа. Двете форми се отличават и по тежестта на протичане на заболяването, като рецесивната форма протича сравнително по-тежко.
Засегнатите лица имат отличителни черти на лицето, като например широко чело, видни и широко раздалечени очи, малък нос и по-широка и с триъгълна форма уста. Взети заедно, тези черти на лицето се описват като "фетални съобщества", защото те приличат на лицето и структурата на развитието на плода.
АР формата се характеризира със скелетни аномалии, включително съкращаване на дългите кости на ръцете и краката, особено на предмишниците; необичайно къси пръсти на ръцете и краката (брахидактилия); клиновидни гръбначни кости, което води до ненормално изкривяване на гръбначния стълб (кифосколиоза); кондензирани или липсващи ребра и нисък ръст.
АД формата има признаци и симптоми, които са сходни, но протичат по-леко. Аномалии на гръбначния стълб и ребрата рядко се наблюдават при автозомно-доминантно форма, и ниския ръст е по-слабо изразен. Характерно е увеличаването на костната минерална плътност (остеосклероза) в допълнение към признаците и симптомите, изброени по-горе. Този вариант се нарича остеосклеротичен.
При АР форма се наблюдават мутации в гена ROR2, на дългото рамо на хромозома 9, който представлява протеин отговорен за развитието на скелета, сърцето и половите органи. При АД форма се наблюдават мутации в гена WNT5A или DVL1, който е отговорен за химичните сигнални пътища. Някои хора с характерните признаци и симптоми на синдрома на Robinow не разполагат с установена мутация в ROR2, WNT5A, или DVL1 ген. В тези случаи, причината на заболяването е неизвестна.
Синдромът на Ръсел-Силвър (Russell Silver Syndrome) се характеризира с бавен растеж, преди и след раждането. Генетичните причини за синдром на Ръсел-Силвър са комплексни. Разстройството често е резултат от ненормално регулиране на някои гени, които контролират растежа. Изследванията са фокусирани върху гени, разположени в определени региони на хромозома 7 и хромозома 11.
Бебетата с това заболяване имат ниско тегло при раждане и често не успяват да растат и да наддават на тегло при очакваната норма (невъзможност да се развиват). Растежът на главата е нормална, обаче, тя може да се окаже твърде голяма в сравнение с останалата част на тялото. Засегнатите деца са слаби и злояди. Някои развиват повтарящи се епизоди на ниска кръвна захар (хипогликемия).
Възрастни със синдром на Ръсел-Силвър са къси; средната височина на засегнатите мъже е около 151 сантиметра и средната височина на засегнатите жени е около 140 сантиметра.
Заболяването се характеризира с: малко триъгълно лице, с високо чело, тясна брадичка, малка челюст, клинодактилия (необичайно извит 5 пръст), неравномерен растеж на някои части на тялото и аномалии на храносмилателната система. Синдромът на Ръсел-Силвър също е свързан с повишен риск от забавено развитие, говорни и езикови проблеми, и когнитивни увреждания.
Повечето случаи на синдром на Ръсел-Силвър са спорадични, което означава, че те възникнат при хора без история на заболяването в семейството им.
Рядко, синдром на Ръсел-Силвър може да се срещне в цели семейства. В някои от засегнатите семейства, условието е да има автозомно доминантен модел на унаследяване. Автозомно-доминантно наследство означава, че едно копие на генетична промяна във всяка клетка е достатъчно да предизвика разстройство. В други семейства, състоянието може да има и автозомно-рецесивен модел на унаследяване. Родителите на дадено лице с автозомно-рецесивно състояние всеки носят едно копие на мутиралия ген, но те обикновено не показват признаци и симптоми на заболяването.
Приемът на калории при децата с RSS трябва да се контролира внимателно, за да се осигури нормален растеж. Ако детето не е в състояние да толерира хранене през устата, може да се използва ентерално хранене с гастростома. Евентуална физиотерапия и оперативни интервенции могат да се приложат.
За да се предотврати утежняващи позата трудности, при деца с разлики в дължината на долните крайници, може да се повиши подметката на тяхната обувка.
Прилага се и терапия с растежен хормон. То може да бъде ефективно, дори когато пациентът не разполага с дефицит на растежния хормон. Лечението с растежен хормон е доказано, за увеличаване на темповете на растеж при пациенти. Това може да даде възможност на детето да започне обучението си в една нормална височина, подобряване на тяхното самочувствие и взаимодействие с други деца.
Синдромът на Секел (Seckel) е рядко наследствено заболяване, характеризиращо се със забавяния на растежа преди раждане (вътрематочно забавяне на растежа), водещо до ниско тегло при раждане. Предава се по автозомно-рецесивен път. Има три варианта на синдрома на Seckel, които включват смущения или промени (мутации) на гени в три различни хромозоми. Първия тип на синдрома Seckel засяга хромозома 3 (3q22-q24); вторият тип засяга хромозома 18 (18p11.31-Q11) и третия тип засяга хромозома 14 (14q21-q22).
Забавеният растеж продължава и след раждане, което води до нисък ръст (нанизъм). Други симптоми и физически характеристики, свързани със синдрома Seckel включват: ненормално малка глава (микроцефалия); различна степен на умствена изостаналост; и / или необичайни черти на лицето, включително "клюноподобна" изпъкналост на носа. Други черти на лицето могат да включват необичайно големи очи, тясно лице, деформирани уши, и / или необичайно малка челюст (микрогнатия). В допълнение, някои от засегнатите деца могат да имат постоянно фиксиране на пръстите в свито положение (клинодактилия), дисплазия на бедрата, дислокация на кост в ръката (радиалната дислокация), и / или други физически аномалии.
Други черепно-лицеви аномалии могат да бъдат необикновено големи очи с полегати клепачни гънки; кривогледство (страбизъм); дисплазия на ушите с отсъстващи части от ухото; и / или по-висок свод на устата, който може да бъде непълно (цепнато небце). В някои случаи, от едната страна на лицето може да се появи по-голям от другия (лицевата асиметрия).Често се наблюдават и аномалии на опорно-двигателната, половата система и хематологични нарушения.
Пренаталната диагноза на синдрома на Секел се прилага чрез фетална ехография. Ниският ръст при синдрома на Seckel включва пропорционален размер на ръцете и краката, което дава възможност за диференциална диагноза със синдроми, които включват нисък ръст и ненормално малки ръце и крака (къси крайници нанизъм).
Синдромът на Смит-Лемли-Опиц (Smith-Lemli-Opitz) е разстройство на развитието, което засяга много части на тялото. Причината за синдрома на Смит са мутации в ген наречен DHCR7. Генът DHCR7 осигурява условия за синтеза на ензим, наречен 7-дехидрохолестерол редуктаза. Този ензим е отговорен за крайния етап в производството на холестерол. Холестеролът е необходим за нормалното ембрионално развитие и има важни функции както преди, така и след раждането. Той е структурен компонент на клетъчните мембрани и е защитна субстанция, покриваща нервните клетки (миелин). Освен това, холестеролът играе роля в производството на някои хормони и храносмилателни киселини. Това състояние се наследява по автозомно рецесивен път. Родителите на дадено лице с автозомно-рецесивно състояние носят по едно копие на мутиралия ген, но те обикновено не показват признаци и симптоми на заболяването.
Това състояние се характеризира с отличителни черти на лицето, малък размер на главата (микроцефалия), умствена недостатъчност или проблеми с обучението, както и поведенчески проблеми. Много засегнати деца имат характерните черти на аутизъм, състояние, което засяга комуникация и социално взаимодействие. Малформации на сърцето, белите дробове, бъбреците, стомашно-чревния тракт и гениталиите са също често срещани. Новородени със синдром на Smith-Lemli-Opitz имат слаб мускулен тонус (хипотония), трудности с храненето и имат склонност към изоставане в растежа. При най-тежко засегнатите лица се наблюдава синдактилия, а някои имат и полидактилия на ръце и крака.
Признаците и симптомите на синдрома на Смит, включващ се в рубриката "Синдроми на вродени аномалии, свързани предимно с нисък ръст", варират в широки граници. Слабо засегнатите лица могат да имат само леки физически аномалии и проблеми с учене и поведението. Тежките случаи могат да бъдат животозастрашаващи и включват дълбоки умствени увреждания и големи физически аномалии.
Коментари към Синдроми на вродени аномалии, свързани предимно с нисък ръст МКБ Q87.1