Бъбречна поликистоза, инфантилен тип МКБ Q61.1
› Какво представлява автозомно-рецесивната бъбречна поликистозна болест
› Диагноза
› Лечение
› Прогноза
Въведение
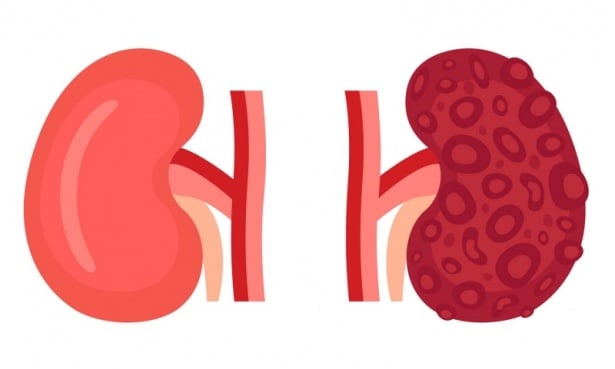
Поликистозата на бъбреците, принадлежаща към групата заболявания кистозна болест на бъбрека, е едно от най-честите животозастрашаващи генетични заболявания. Това нелечимо наследствено заболяване се характеризира с образуването на изпълнени с течност кисти в бъбреците на засегнатите индивиди. Тези кисти се увеличават с течение на времето.
Първоначално се е смятало, че кистите причиняват бъбречната недостатъчност. Сега се счита, че увреждането на бъбреците, установено при поликистоза, е всъщност резултат от имунната система на организма. Имунната система в опитите си да премахне кистите от бъбреците, постепенно разрушава здравата бъбречна тъкан.
Здравият бъбрек е приблизително със същия размер като човешки юмрук. Информация за анатомията на бъбреците може да прочетете тук:
Кистите при това заболяване, които могат да бъдат толкова малки, колкото глава на карфица, или по-големи от грейпфрут, могат да уголемят бъбреците. Съществуват два вида бъбречна поликистоза - инфантилен тип, който в повечето случаи показва симптоми преди раждането, и адулторен тип.
Бъбречната поликистоза се експресира както по рецесивен, така и по доминантен модел. Рецесивното унаследяване няма да причини заболяване при деца, освен ако не се предаде генетично и от двамата родители. Доминантното унаследяване може да бъде предадено генетично само от единия родител. Индивидите, засегнати от автозомно-доминантната форма на бъбречна поликистоза, имат много по-често срещания вид - адулторен тип поликистоза на бъбреците. Индивидите с автозомно-рецесивна форма на бъбречна поликистоза са с инфантилния вид.
Бъбречна поликистоза, адулторен тип, е много по-често срещана. Времето и степента на появата на симптомите на тази форма бъбречна поликистоза може да варира значително. Признаците на заболяването при адулторния вид обикновено започват да се появяват на възраст между 20 и 50 години. Органното увреждане прогресира по-бавно при адулторната форма на бъбречна поликистоза, отколкото при инфантилната форма.
Повече информация за бъбречна поликистоза от адулторен вид може да прочетете тук:
» Бъбречна поликистоза, адулторен тип мкб Q61.2
» Кистозна болест на бъбрека мкб Q61
Какво представлява?
Автозомно-рецесивната бъбречна поликистозна болест е рядко наследствено заболяване в детството. Въпреки че е рядко състояние, това е един от най-честите бъбречни проблеми, които засягат малки деца, и най-честото наследствено кистозно заболяване на бъбреците, което се среща в ранна и детска възраст. Смята се, че около 1 на 20 000 бебета се раждат с това състояние. Двата пола са засегнати еднакво. Различава се от автозомно-доминантна бъбречна поликистозна болест (адулторен тип), която обикновено се среща при по-възрастна популация.
Заболяването се свързва с група вродени фиброкистозни синдроми. Клиничният спектър показва широка вариабилност, варираща от перинатална смърт до по-леко прогресивна форма, която може да не бъде диагностицирана до юношеството. Рисковите фактори за по-тежка форма на заболяването при кърмачета са олигохидрамнион или анхидрамнион, пренатално уголемяване на бъбреците и необходимостта от постнатално поддържане на дишането
Признаците на автозомно-рецесивната бъбречна поликистозна болест често започват преди раждането, така че често се нарича бъбречна поликистоза, инфантилен тип, но някои хора развиват симптоми едва по-късно в детството или дори в зряла възраст.
Индивидите, засегнати от бъбречна поликистоза, инфантилен тип, често са мъртвородени. Около 30% от новородените с автозомно-рецесивната бъбречна поликистозна болест умират през първата седмица от живота си. Сред живородените засегнати бебета с инфантилен тип поликистоза на бъбреците много малко от тези деца оцеляват до 2-годишна възраст.
Етиология
Бъбречна поликистоза, инфантилен тип, се причинява от генетична мутация в участъка на късото рамо на хромозома 6 (6p21). Мутиралият ген PKHD1 кодира мембранно свързан рецептор-подобен протеин, наречен фиброцистин. Този протеин участва в цилиарната сигнализация, необходима за регулиране на пролиферацията и диференциацията на епителните клетки на бъбречните и жлъчните пътища. В случаи, при които генната мутация на фиброцистина води до по-малко тежки дефекти, може да бъде налична достатъчна бъбречна функция, за да оцелее бебето.
И двамата родители трябва да са носители на тази мутация, за да бъдат засегнати техните деца от заболяването. В случай на двама родители носители вероятността детето да бъде засегнато е 25%.
Ако само единият родител носи мутиралия ген, детето няма да получи заболяването, въпреки че детето може да получи генна мутация. Детето е „носител“ на заболяването и може да предаде генната мутация на следващото поколение.
Патофизиология
Заболяването е резултат от мутация в местоположението на гена PKHD1 (поликистозно бъбречно и чернодробно заболяване) върху хромозома 6p. Това води до двустранно симетрично микрокистозно заболяване, което се появява в дисталните извити тубули и събирателните канали. Броят на засегнатите канали определя възрастта на клинична проява.
- Перинатален тип: най-честият тип на заболяването
- олигохидрамнион и белодробна хипоплазия
- 75% от случаите с летален изход в рамките на 24 часа след раждането
- минимална чернодробна фиброза
- Неонатален тип: минимална чернодробна фиброза
- Инфантилен тип: умерена перипортална фиброза
- Ювенилен тип: груба чернодробна фиброза
- портална хипертония със спленомегалия и портосистемни варици
- Болест на Кароли
- Вродена чернодробна фиброза: степента на която е обратно пропорционална на възрастта на представяне
- Множество жлъчни хамартоми
Клинични характеристики
Бъбреците са засегнати двустранно и по време на бременността обикновено има олигохидрамнион поради увредена бъбречна функция и невъзможност да се образува значителни количества фетална урина.
Най-значимото последствие от олигохидрамниона е белодробна хипоплазия, поради което новородените не разполагат с достатъчно капацитет на белия дроб, за да оцелеят.
Бъбреците са значително увеличени и са с тенденция да изпълнят ретроперитонеума и да изместят абдоминалното съдържание. Те са с тенденция да се уголемяват симетрично.
Бебе, родено с инфантилна форма на бъбречна поликистоза, има много широки, ниско поставени уши, остър нос, малка брадичка и епикантални гънки. Големи, твърди маси могат да се палпират на задната страна на двата хълбока. Бебето обикновено има проблеми с дишането.
Тежестта на заболяването варира, като признаците често започват още преди раждането. При най-тежките случаи смърт настъпва часове или дни след раждането поради дихателни затруднения или недостатъчност. Някои индивиди с автозомно-рецесивната форма на бъбречна поликистоза развиват симптоми едва в по-късно детство и дори в зряла възраст. Заболяването обикновено засяга черния дроб и далака, което води до нисък брой на кръвните клетки, разширени вени и хемороиди.
Дете с това заболяване на възраст 5-10 години е с тенденция да развие високо налягане в кръвоносните съдове, които свързват червата и черния дроб (портална система). Освен високо кръвно налягане, децата страдат от инфекции на пикочните пътища и често уриниране. Тъй като бъбречната функция е от решаващо значение за ранното физическо развитие, децата с автозомно-рецесивно унаследена поликистоза на бъбреците обикновено са по-малки от средния размер за възрастта.
Усложнения
Децата с автозомно-рецесивната бъбречна поликистозна болест, които преживяват раждането, често имат проблеми с бъбреците и черния дроб, които могат да повлияят на дишането им.
- Проблеми с дишането - най-тежките случаи често умират часове или дни след раждането, тъй като не могат да дишат достатъчно добре, за да живеят. Белите им дробове не се развиват както трябва в утробата.
- Бъбречна недостатъчност - децата, родени с това заболяване, често развиват бъбречна недостатъчност, преди да достигнат зряла възраст.
- Проблеми с черния дроб - фиброза на черния дроб се появява във всички случаи на заболяването и обикновено още при раждането. Фиброзата може да доведе до намалена чернодробна функция и други чернодробни проблеми. Проблемите с черния дроб при автозомно-рецесивната бъбречна поликистозна болест са с тенденция да стават все по-притеснителни с течение на времето.
- Високо кръвно налягане - повечето деца с това заболяване имат високо кръвно налягане. Високото кръвно налягане увеличава риска от сърдечни заболявания и инсулт, както и може допълнително да увреди бъбреците на детето.
В прогресията на заболяването се развива чернодробна недостатъчност и хронична бъбречна недостатъчност. Двустранното кистозно заболяване на бъбреците е тежко и трудно съвместимо с живота.
Диагноза
Диагнозата се поставя въз основа ултразвуково изследване на плода или новороденото, което разкрива уголемени бъбреци с анормален вид, но големи кисти, като например тези в автозомно-доминантно унаследената бъбречна поликистоза рядко се виждат. Ултразвуково изследване на черния дроб също помага на диагностиката.

Изображение: radiopaedia.org, CC BY-NC-SA 3.0
Ултразвуковото изследване е най-полезният образен метод за диагностициране на заболяването, но често е необходима допълнителна информация, за да се потвърди диагнозата (например фамилна анамнеза, ултразвуково изследване на родителите, клиничен преглед и генетичен анализ).
Изследване с ядрено-магнитен резонанс и магнитно-резонансна холангиопанкреатография се провежда при пациенти с клинични усложнения на чернодробно заболяване.
При деца с вече диагностицирана автозомно-рецесивната бъбречна поликистозна болест се препоръчва ежегодно изследване на корема за проследяване на признаци на портална хипертония. При тежко засегнати кърмачета с прогресиращо заболяване размерът на бъбреците трябва да се проследява според клиничните нужди.
Диференциална диагноза
Диференциалната диагноза на това състояние включва:
- Гломерулокистозна бъбречна болест
- Автозомно-доминантна поликистозна бъбречна болест
- Дифузна кистозна дисплазия
- Синдром на Бекуит-Видеман (Beckwith-Wiedemann)
- Синдром на Лорънс-Мун-Бейдл (Laurence-Moon-Beidl)
- Бъбречна дисплазия, свързана с тризомия 13
Лечение
Не е известно лечение за вроденото поликистозно заболяване на бъбреците. Случаите на инфантилна форма на бъбречна поликистоза често завършват летално преди навършване на 2-годишна възраст.
Терапията при бъбречна поликистоза, инфантилен тип, е поддържаща - медикаменти за контрол на високото кръвно налягане и антибиотици за лечение на инфекциите на пикочните пътища. Приемът на повишени количества питателна храна подобрява растежа при деца с автозомно-рецесивен тип поликистоза на бъбреците. В някои случаи се използват растежни хормони. В отговор на бъбречната недостатъчност се прилага диализа или трансплантация. Ако се развие сериозна чернодробна заболеваемост, някои хора могат да претърпят комбинирана чернодробна и бъбречна трансплантация.
Прогноза
Прогнозата като цяло е лоша, но също така може да варира значително в зависимост от тежестта на състоянието. Около 40% (диапазон 30-50%) от засегнатите бебета умират по време на перинаталния период поради белодробна хипоплазия и белодробна недостатъчност.
Ако рутинните изследвания установят проблеми с бъбреците по време на бременност, бебето обикновено ще има по-лоша прогноза от дете, което е диагностицирано на по-късен етап.
Но като цяло автозомно-рецесивната бъбречна поликистозна болест е тежко състояние и около 1 от 3 бебета ще умрат от тежки затруднения с дишането през първите 4 седмици след раждането. Около 8 или 9 от 10 бебета със заболяването, които оцелеят през първия месец от живота, ще живеят до поне 5 години.
Трудно е да се предвиди точно колко дълго ще живее дете с бъбречна поликистоза, инфантилен тип, тъй като има много малко данни, показващи дългосрочните нива на преживяемост. Но с напредъка в лечението и по-доброто разбиране на състоянието, все по-голям брой деца със заболяването живеят добре в зряла възраст.
Заглавно изображение: freepik.com
Библиография
https://www.niddk.nih.gov/health-information/kidney-disease/polycystic-kidney-disease/autosomal-recessive-pkd
https://rarediseases.info.nih.gov/diseases/8378/autosomal-recessive-polycystic-kidney-disease
https://emedicine.medscape.com/article/377154-overview
https://en.wikipedia.org/wiki/Autosomal_recessive_polycystic_kidney_disease
https://www.mayoclinic.org/diseases-conditions/polycystic-kidney-disease/symptoms-causes/syc-20352820
https://radiopaedia.org/articles/autosomal-recessive-polycystic-kidney-disease
ЗАБОЛЯВАНЕТО е свързано към
- Отделителна система
- Алтернативно лечение на бъбречни кисти
- Q61 Кистозна болест на бъбрека
- Q61.2 Бъбречна поликистоза, адулторен тип
- Високоспециализирана операция на голяма бъбречна киста в МБАЛ „Княгиня Клементина“
- Q61.0 Вродена единична киста на бъбрека
- Q61.3 Бъбречна поликистоза, неуточнена
- Q61.5 Медуларна бъбречна киста
- Според научно откритие бъбреците работят и като помпа при филтрирането на кръвта
Коментари към Бъбречна поликистоза, инфантилен тип МКБ Q61.1