Гломерулни увреждания при други болести на ендокринната система, разстройства на храненето и на обмяната на веществата МКБ N08.4
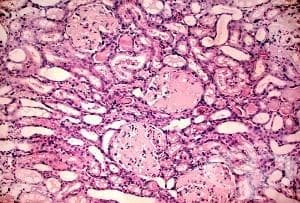
Бъбречните гломерули могат да се увредят при различни заболявания.
Това може да се наблюдава при инфекция, различни токсични нокси, ензимни дефекти и други.
Клиничната изява при увреждане на бъбречните гломерули може да бъде разнообразна.
Към гломерулни увреждания при други болести на ендокринната система, разстройства на храненето и на обмяната на веществата спадат тези при амилоидоза, болест на Fabry (-Anderson) и недоимък на лецитин-холестерол-ацилтрансфераза.
Към гломерулни увреждания при други болести на ендокринната система, разстройства на храненето и на обмяната на веществата принадлежи амилоидозата.
Амилоидозата представлява абнормалното натрупване на протеин (амилоид) в междуклетъчното пространство на тъканите и органите (включително и на бъбречните гломерули).
Етиологията за развитието на амилоидоза не е изяснена, но е установена известна зависимост от възрастта (среща се по-често над 40 години), пола (по-често при мъже) и фамилната обремененост.
Клиничната изява на бъбречната амилоидоза протича протрахирано с поява на белтък в урината (протеинурия), артериална хипертония и постепенно прогресира към развитието на хронична бъбречна недостатъчност. При отделяне на голямо количество белтък с урината се развиват хипопротеинемични отоци по лицето и крайниците, събиране на течност в коремната и в плевралната кухина. Слезката и езикът обикновено са с увеличени размери поради натрупания амилоид.
При подозиране на диагнозата и фамилна обремененост може да се извърши ДНК - типизиране или да се извърши бъбречна биопсия. Хистологичната картина на амилоидозата е характерна: виждат се плътни амилоидни отлагания в бъбречните гломерули.
Поради напредващата бъбречна недостатъчност единственото радикално лечение на бъбречната амилоидоза е трансплантацията на здрав бъбрек. Паралелно трябва да се поддържа нормалната хомеостаза на организма, като се приемат медикаменти за понижаване на кръвното налягане, диуретици и други.
Повече за амилоидозата можете да прочетете тук.
Към гломерулни увреждания при други болести на ендокринната система, разстройства на храненето и на обмяната на веществата принадлежи болестта на Фабри (Fabry).
Болестта на Фабри (Fabry) представлява рядък Х-свързан генетичен дефект (около 1: 40 000 души), при който липсва ензимът алфа-галактозидаза А. Поради дефицита му в организма се натрупват сфинголипиди. Наблюдава се натрупването им във всички органи и тъкани (включително и бъбреците), което предизвиква оток и разрастване на ендотелни клетки.
Клиничната изява на болестта на Фабри започва още от ранно детство. Детето се оплаква от дифузна болка по крайниците и в корема, появява се белтък в урината, артериална хипертония, сърцебиене, прескачане на сърцето, аритмия, катаракта, генерализирана слабост и липса на апетит. Могат да се наблюдават кожни промени под формата на ангиокератоми (малки, неболезнени папули), анхидроза (липса на потене) или точно обратното - хиперхидроза (засилено потене) на даден участък.
За поставяне на диагнозата е необходимо да се снеме добра фамилна анамнеза и да се тестува активността на галактозидазната активност. Попнякога е необходимо да се извърши и бъбречна биопсия.
За лечение на болест на Фабри се използва заместителна терапия с изкуствени ензими- Replagal и Fabrazyme. При тежко бъбречно или чернодробно увреждане пациентът трябва да се подготви за трансплантация.
Повече за болест на Фабри можете да прочетете тук.
Към гломерулни увреждания при други болести на ендокринната система, разстройства на храненето и на обмяната на веществата принадлежи недоимъкът на лецитин-холестерол-ацилтрансфераза.
Недоимъкът на лецитин-холестерол-ацилтрансфераза също е ензимен дефект. При липсата на този ензим свободният холестерол не може да се метаболизира до холестеролови естери. Така свободният холестерол се отлага в различни тъкани и органи (включително и бъбреците).
Клиничната изява на недоимъкът на лецитин-холестерол-ацилтрансфераза включва катаракта, хемолитична анемия, протеинурия, артериална хипертония и прогресираща бъбречна недостатъчност.
Лабораторните промени при това заболяване се характеризират с много ниско ниво на HDL - и свободния холестерол, високи триглицериди и ниско ниво на холестероловите естери. За да се постави диагнозата със сигурност е необходимо да се направи генетичен анализ.
Лечението при дефицита на лецитин-холестерол-ацилтрансфераза е предимно симптоматично- третиране на артериалната хипертония, анемията и бъбречната недостатъчност.
Повече за дефицит на лецитин-холестерол-ацилтрансферазата можете да прочетете тук.
Коментари към Гломерулни увреждания при други болести на ендокринната система, разстройства на храненето и на обмяната на веществата МКБ N08.4