Чиста хиперхолестеролемия МКБ E78.0

Към рубриката чиста хиперхолестеролемия се включва група от заболявания, към които принадлежи фамилна хиперхолестеролемия.
Фамилната хиперхолестеролемия е генетично заболяване, характеризиращо се с високи нива на холестерол, особено много високи нива на липопротеини с ниска плътност (LDL, "лош холестерол"), в кръвта и ранни сърдечно-съдови заболявания. Най-честите мутации намаляват броя на функционалните LDL рецептори в черния дроб. Фамилната хиперхолестеролемия е класифицирана като фамилна дислипидемия тип 2. Има пет типа фамилна дислипидемия (без да се включват подвидовете) и всеки от тях се класифицира както от променения липиден профил, така и от генетичната аномалия. Например, високият LDL (често поради LDL рецепторен дефект) е тип 2. Други включват дефекти в метаболизма на хиломикрон, метаболизма на триглицеридите и метаболизма на други частици, съдържащи холестерол, като липопротеини с много ниска плътност (VLDL) и липопротеин със средна плътност (IDL).
Фамилната хиперхолестеролемия е интересно заболяване, върху което вниманието е насочено от патолог Harbitz, поради съвпадението на липидни отлагания (xanthomata) в сухожилията и внезапна смърт, преди да бъде описано от Muller като клинична единица през първата половина на миналия век. Проучвания при пациенти и култивирани клетки разкриват дефект в рецептора за LDL и завършват с Нобеловата награда за Браун и Голдщайн, които също изследват регулирането на синтетичния път на холестерола.
Класификация
Фамилната хиперхолестеролемия може да бъде класифицирана в „хетерозиготен“ и „хомозиготен“ клиничен фенотип. Хетерозиготният фенотип се наследява по автозомно доминиращ начин и има концентрация на LDL-холестерол между 5 и 12 милимола/литър, ксантоматите на сухожилията са често срещани, а коронарна болест на сърцето преди 55-годишна възраст също е често срещано явление. Водещата причина за хетерозиготния фенотип е мутация в LDL рецептора.
Хомозиготният фенотип има по-тежка LDL хиперхолестеролемия (над 12 милимола/литър) и обикновено има и ксантомати на кожата и сухожилията. Генетичните причини за хомозиготния фенотип включват съвпадение на два LDL рецепторни дефекта, като се унаследява по рецесивен модел.
По този начин терминът фамилна хиперхолестеролемия е ограничен до специфични нарушения и не означава просто наследствена хиперхолестеролемия. Докато някои липидолози все още могат да запазят термина фамилна хиперхолестеролемия специално за дефекти на LDL рецептора, хетерозиготният фенотип е бил и все още се използва за идентифициране на субекти, за които се предполага, че имат дефекти на LDL рецептор, но които може да са имали някои от другите дефекти.
Епидемиология
Глобалното разпространение на заболяването е приблизително 10 милиона души. В повечето изследвани популации хетерозиготният фенотип се среща при около 1:250 души, но не всички развиват симптоми. Хомозиготният фенотип се появява за около1:1 000 000.
Мутациите на LDL рецепторите са по-чести в определени популации, вероятно поради генетичен феномен, известен като основателски ефект - те са основани от малка група индивиди, един или няколко от които са носители на мутацията. Африканците, френските канадци, ливанските християни и финландците имат високи нива на специфични мутации, които правят фамилната хиперхолестеролемия особено често срещана в тези групи.
Етиология
Най-честите генетични дефекти при фамилна хиперхолестеролемия са мутации на LDL рецепторите (разпространение 1 на 250, в зависимост от популацията), аполипопротеин В (ApoB) мутации (разпространение 1 на 1000), пропротеин конвертаза субтилизин/кексин тип 9 мутации (по-малко от 1 на 2500) и ген ARH.
LDL рецептор
Генът за LDL рецептор е разположен на късото рамо на хромозома 19. Съдържа 18 екзона, а протеиновият генен продукт съдържа 839 аминокиселини в зряла форма. Едно анормално копие (хетерозигот) на фамилна хиперхолестеролемия причинява сърдечно-съдови заболявания до 50-годишна възраст в около 40% от случаите. Наличието на две анормални копия (хомозигота) причинява ускорена атеросклероза в детска възраст, включително нейните усложнения. Нивата на LDL в плазмата са обратно свързани с активността на LDL рецептора (LDLR). Хомозиготите имат LDLR активност под 2%, докато хетерозиготите имат дефектна LDL обработка с рецепторна активност 2-25%, в зависимост от естеството на мутацията. Известни са над 1000 различни мутации.
Има пет основни класа заболяването поради LDLR мутации:
- Клас I: LDLR изобщо не се синтезира.
- Клас II: LDLR не се транспортира правилно от ендоплазмения ретикулум до апарата на Голджи за експресия на клетъчната повърхност.
- Клас III: LDLR не свързва правилно LDL на клетъчната повърхност поради дефект в аполипопротеина B100 или в LDL-R.
- Клас IV: LDLR, свързан с LDL, не се групира правилно в покрити с клатрин ямки за рецептор-медиирана ендоцитоза.
- Клас V: LDLR не се групира обратно към клетъчната повърхност.
Аполипопротеин В
Аполипопротеин В, във формата си ApoB 100, е основният аполипопротеин или протеинова част на липопротеиновата частица. Неговият ген е разположен на втора хромозома. Фамилната хиперхолестеролемия често се свързва с мутацията на R3500Q, която причинява заместване на аргинин с глутамин в позиция 3500. Мутацията е разположена върху част от протеина, която обикновено се свързва с LDL рецептора, и свързването намалява в резултат на мутацията. Подобно на LDLR, броят на анормалните копия определя тежестта на хиперхолестеролемията.
Пропротеин конвертаза субтилизин/кексин тип 9
Мутациите в гена на пропротеин конвертаза субтилизин/кексин тип 9 (PCSK9) са свързани с автозомно доминиращ (изисква само едно анормално копие) в доклад от 2003 година. Генът е разположен в първата хромозома и кодира аминокиселинен протеин, който се експресира в черния дроб. Предполага се, че PCSK9 причинява заболяването главно чрез намаляване на броя на LDL рецепторите в чернодробните клетки.
Ген ARH
Аномалии в гена ARH са докладвани за първи път в семейство през 1973 година. За разлика от другите причини, за развитие на фамилна хиперхолестеролемия са необходими две анормални копия на гена (автозомно-рецесивен). Мутациите в протеина са склонни да причиняват производството на съкратен протеин. Истинската му функция е неясна, но изглежда, че играе роля в отношенията между LDL рецептора и покритите с клатрин ямки. Хората с автозомно-рецесивна хиперхолестеролемия са склонни да имат по-тежко заболяване от LDLR-хетерозиготите, но по-малко тежко от LDLR-хомозиготите.
Патофизиология
Липопротеините с ниска плътност обикновено циркулират в тялото в продължение на 2,5 дни и впоследствие частта от LDL холестерол от аполипопротеин В се свързва с LDL рецептора на чернодробните клетки, предизвиквайки неговото усвояване и храносмилане. Този процес води до отстраняване на LDL от кръвоносната система. Синтезът на холестерол от черния дроб се потиска по пътя на HMG-CoA редуктазата. При фамилна хиперхолестеролемия функцията на LDL рецептор е намалена или липсва, и LDL циркулира със средна продължителност от 4,5 дни, което води до значително повишено ниво на LDL холестерол в кръвта с нормални нива на други липопротеини. При мутации на ApoB, намаленото свързване на LDL частици с рецептора причинява повишеното ниво на LDL холестерол. Не е известно как мутацията причинява дисфункция на LDL рецептора при мутации на PCSK9 и ARH.
Изображение: freepik.com
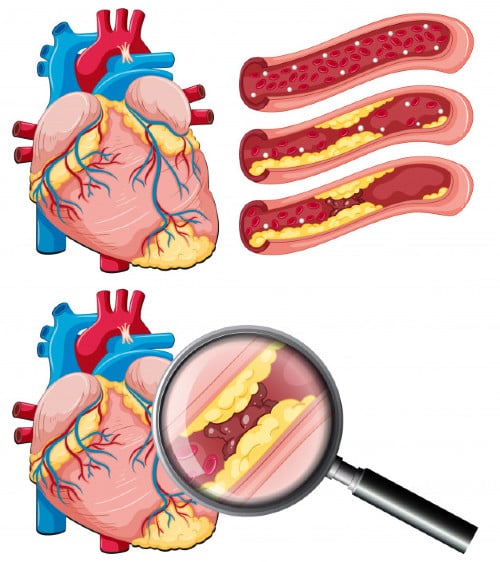
Въпреки че атеросклерозата се среща до известна степен при всички хора, хората с фамилна хиперхолестеролемия могат да развият ускорена атеросклероза поради излишното ниво на LDL. Степента на атеросклероза приблизително зависи от броя на LDL рецепторите, които все още са изразени, и от функционалността на тези рецептори. При много хетерозиготни форми на фамилна хиперхолестеролемия рецепторната функция е само леко нарушена и нивата на LDL ще останат относително ниски. При по-сериозните хомозиготни форми рецепторът изобщо не се експресира.
Клинична картина
В клиничната практика най-често се среща хетерозиготен фенотип на заболяването, докато рядко срещаният хомозиготен фенотип има много по-лоши последици. Пациентите с фамилна хиперхолестеролемия обикновено са имали исхемична болест на сърцето в миналото, но днес с по-голяма осведоменост, по-прости химически тестове, които са широко достъпни, диагнозата се поставя по-рано.
Фамилната анамнеза за преждевременна исхемична болест на сърцето е много полезна за идентифициране на автозомно доминиращ начин на унаследяване и произходът на пациента може да предполага молекулярен дефект, ако има връзка към популация с основополагащ ефект. Въпреки високия риск от сърдечни заболявания при хетерозиготен фенотип, рискът е значително модифициран от други гени и фактори на околната среда, така че фамилната анамнеза понякога може да бъде подвеждащо доброкачествена. В такава обстановка трябва да се обмисли рецесивно разстройство или да се обмисли нова мутация, водеща до доминиращо унаследен модел.
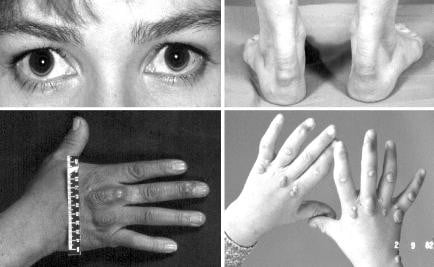
Физическите признаци, дължащи се на отлагането на липиди в окото, кожата и сухожилията, са ценни, но не неизменни признаци. Физическите признаци са изобразени на снимката по-долу. Хетерозиготният фенотип обикновено има сухожилни прояви сам и хомозиготният фенотип има тенденция да има както сухожилни, така и кожни прояви. При хетерозиготния фенотип проявите обикновено не се забелязват лесно на възраст между 25 и 30 години, докато при хомозиготния фенотип те обикновено са очевидни преди навършване на 10 години.
Изображение: ncbi.nlm.nih.gov
Палпацията на ахилесовото сухожилие изисква известен опит, но е най-добрият клиничен признак за диагностициране на фамилна хиперхолестеролемия. Ултразвуковото изследване може да установи по-точни размери. Удебеляването на сухожилията чрез отлагане на холестерол може да доведе до възпаление и болка и в крайна сметка отлагането на липиди води до неправилна повърхност с възли и понякога видимо разширени сухожилия. Предполага се, че високото напрежение, предавано през относително аваскуларното сухожилие на неговите прегради, съдържащи кръвоносните съдове, причинява ексудация или кървене в тъканта. Изглежда, че съединителната тъкан свързва липопротеините и холестеролът се натрупва. Нечесто ксантоматите също се образуват в екстензорните сухожилия на пръстите и рядко другаде в тялото. Откриването на ксантомати при клиничен преглед има не само диагностични последици, но може също така да показва риск, тъй като е установено, че пациентите с фамилна хиперхолестеролемия с ксантомати имат повече сърдечни заболявания.
Отлагането на липиди върху роговицата на окото води до бяла линия под формата на полумесец, наричана arcus cornealis. Отлагането на липиди в клепачите като плоски жълти плаки, наричано ксантелазма, не е специфично за фамилна хиперхолестеролемия и дори може да се наблюдава при лица без значителни нарушения на плазмените липопротеини.
Съдова болест
Субклиничната атеросклероза е очевидна при млади хора с фамилна хиперхолестеролемия, когато се търси чрез съдови образни техники. При хомозиготни пациенти с аортната стеноза е добре установено усложнение в резултат на отлагане на липиди в аортната клапа. Други съдове също могат да имат турбулентен поток при хомозиготен фенотип, особено бедрените артерии.
Атеросклерозата се разбира несъвършено, но очевидно е многофакторно заболяване, при което особено при LDL хиперхолестеролемията играе силна роля. При фамилна хиперхолестеролемия е по-тежко и води до клинични усложнения по-рано. Високите концентрации на LDL, пребиваващи в циркулацията за продължителни периоди, позволяват по-окислителна модификация в циркулацията и тъканите. Тези окислени липопротеини оказват неблагоприятно въздействие върху съдовия ендотел, насърчавайки привличането на моноцити, които проникват в или остават в субендотелното пространство, където се наблюдава индолентна форма на хронично възпаление.
Диагноза
Приблизително 85% от хората с това разстройство не са диагностицирани и следователно не получават лечение за понижаване на липидите. Констатациите от физикалния преглед могат да помогнат на лекаря да постави диагнозата фамилна хиперхолестеролемия. Ксантомите на сухожилията се наблюдават при 20-40% от хората с това заболяване и са патогномонични за състоянието. Може да се наблюдава и ксантелазма или аркус на роговицата. Тези общи признаци подкрепят диагнозата, но са неспецифични констатации.
Лабораторни изследвания
Диагнозата както на хомозиготен, така и на хетерозиготен фенотип се основава главно на откриването на тежки повишения на LDL холестерол (LDLc) при липса на вторични причини за хиперхолестеролемия с нива на триглицеридите, които са в референтния диапазон или леко повишени и нива на HDL холестерол (HDLc), които са в рамките на референтната стойност обхват или леко нисък. Вероятна диагноза на хетерозиготен фенотип може да бъде поставена, ако нивото на LDLc е по-голямо от 330 милиграма/децилитър или ако има ксантоми на сухожилията при пациент с ниво на LDLc над 95-ия процентил. Окончателната диагноза може да бъде поставена само с анализ на ген или рецептор.
Значително повишаване на серумните нива на триглицеридите трябва да повиши възможността за друго липидно разстройство.
Нивата на холестерола са силно повишени при деца и възрастни с хомозиготен фенотип, като общите нива на холестерола и LDLc са над 600 милиграма/децилитър и нивата на триглицеридите в референтния диапазон.
При пациенти с хетерозиготен фенотип нивата на LDLc обикновено са по-високи от 250 милиграма/децилитър и обикновено се увеличават с възрастта. Нивото на LDLc по-високо от 200 милиграма/децилитър при пациент на възраст под 20 години силно предполага хетерозиготен фенотип на заболяването или, вероятно, фамилен лиганд с дефект apoB-100. При възрастни нивата на LDLc по-високи от 290-300 милиграма/децилитър предполагат хетерозиготен фенотип.
При липса на симптоми или признаци, предполагащи конкретно разстройство, трябва да се извърши ограничена обработка, за да се изключи вторичната хиперхолестеролемия. Основните тестове за изключване на диабет, хипотиреоидизъм, чернодробни заболявания и бъбречни заболявания обикновено са достатъчни.
Образни изследвания
Пациентите с хомозиготен фенотип трябва подлежат на доплер ехокардиографска оценка на сърцето и аортата ежегодно, компютърно-томографска коронарна ангиография на всеки 5 години или по-често, ако е клинично показано, като се вземе предвид облъчването и тежестта на субклиничното заболяване.
Деца с хомозиготен фенотип трябва да бъдат насочени към детски кардиолог за разглеждане на съдови образни изследвания, които могат да насочат лечението на хиперхолестеролемия.
Рентгенографското изображение на ахилесовото сухожилие помага точно да се измери ксантома на ахилесовото сухожилие.
Други тестове
Липопротеиновата електрофореза е скъпа и е ненужна за диагностицирането на заболяването. Освен това, при липса на препаративно ултрацентрифугиране, то няма място в обработката на липидно разстройство. Ако анализът на липидите на гладно разкрие повишени нива на триглицериди и диагнозата е под въпрос, бета количествено определяне (ултрацентрифугиране и електрофореза) може да се извърши.
Анализът на LDL рецепторите може да се използва за идентифициране на специфичния дефект на LDL рецептора. Този анализ обаче може да се извършва само в определени изследователски лаборатории и е скъп.
Лечение
Съвременната фармакотерапия може да постигне желани концентрации на LDL в хетерозиготи, но лечението на хомозиготи остава проблематично.
Хетерозиготен фенотип
Фамилната хиперхорестеролемия обикновено се лекува със статини. Статините действат чрез инхибиране на ензима хидроксиметилглутарил CoA редуктаза (HMG-CoA-редуктаза) в черния дроб. В отговор черният дроб произвежда повече LDL рецептори, които премахват циркулиращия LDL от кръвта. Статините ефективно понижават нивата на холестерола и LDL, въпреки че понякога се изисква допълнителна терапия с други лекарства, като секвестиращи жлъчни киселини (холестирамин или колестипол), препарати от никотинова киселина или фибрати. Необходим е контрол на други рискови фактори за сърдечно-съдови заболявания, тъй като рискът остава малко повишен, дори когато нивата на холестерола се контролират. За разлика от останалата част от населението, хората с чиста хиперхолестеролемия имат високи нива на холестерол от раждането си, вероятно увеличавайки относителния им риск. Преди въвеждането на статините, клофибрат (по-стар фибрат, който често причинява камъни в жлъчката), пробукол (особено при големи ксантоми) и тироксин са били използвани за намаляване на нивата на LDL холестерол.
Алирокумаб и еволокумаб, и двете моноклонални антитела срещу PCSK9, са специално посочени като допълнение към диетата и максимално поносима терапия със статини за лечение на възрастни с хетерозиготна фамилна хиперхолестеролемия, които изискват допълнително понижаване на LDL холестерола.
Хомозиготен фенотип
Хомозиготният вариант на е по-труден за лечение. LDL (липопротеините с ниска плътност) рецептори са минимално функционални, ако изобщо са. Само високите дози статини, често в комбинация с други лекарства, са умерено ефективни за подобряване на нивата на липидите. Ако медицинската терапия не е успешна при намаляване на нивата на холестерола, може да се използва LDL афереза - метод на лечение, при който се филтрира LDL от кръвния поток в процес, напомнящ диализа.
За трансплантация на черен дроб могат да се обмислят много тежки случаи. Това осигурява на черния дроб нормално функциониращи LDL рецептори и води до бързо подобряване на нивата на холестерола, но с риск от усложнения от трансплантация на твърди органи (като отхвърляне, инфекции или странични ефекти на лекарството, необходимо за потискане на отхвърлянето). Други хирургични техники включват частична хирургия на илеалния байпас, при която част от тънките черва се байпасира, за да се намали абсорбцията на хранителни вещества и оттам холестерол, и портакавална шънтова хирургия, при която порталната вена е свързана с кухата вена, за да позволи на кръвта с хранителни вещества от червата, за да заобиколи черния дроб.
Evinacumab, моноклонално антитяло, инхибиращо ангиопоетиноподобен протеин 3, е одобрен през 2021 година за допълнителна терапия.
Деца
Като се има предвид, че фамилна хиперхорестеролемия присъства от раждането и атеросклеротичните промени могат да започнат в началото на живота, понякога е необходимо да се лекуват юноши или дори тийнейджъри със средства, които първоначално са разработени за възрастни. Поради съображения за безопасност, много лекари предпочитат да използват секвестранти на жлъчна киселина и фенофибрат, тъй като те са разрешени за деца. Независимо от това, статините изглеждат безопасни и ефективни, и при по-големи деца могат да се използват както при възрастни.
Заглавно изображение: freepik.com
Симптоми и признаци при Чиста хиперхолестеролемия МКБ E78.0
- Сърдечно заболяване
- Подкожни подутини
- Жълтеникави петна около клепачите
- Ксантоматозна кожа
- Туберозен ксантом
- Ксантоми под кожата
Продукти свързани със ЗАБОЛЯВАНЕТО
СУОНСЪН БЕРБЕРИН капсули 400 мг * 60
ПРОМО
БИЛКА ЧУДОДЕЙКА ТИНКТУРА ЧИСТИ АРТЕРИИ 100 мл

ЦЯЛОСТЕН КОМПЛЕКС ФИБРИ С ФРУКТООЛИГОЗАХАРИДИ (F.O.S.) капсули * 90 VIRIDIAN

ХОЛЕСТЕРОЛ КЛИЙНЪР капсули * 60

ЕНТЕРОСАН ФОРТЕ таблетки 360 мг * 20
Термолабилни
НОРМОЛИП 5 капсули * 30

ПЮР НУТРИШЪН ОМЕГА - 3 РИБЕНО МАСЛО 180 EPA / 120 DHA дражета * 100

СУОНСЪН ПЪЛЕН СПЕКТЪР ОТ ЧЕСЪН капсули 400 мг * 60
ПРОМО
СОРТИС таблетки 40 мг * 30
ФИБРАНОР капсули 200 мг * 30 ТЕВА

ХОЛЕСТЕМИН капсули * 30 ДОЛЕРАН ФАРМА

АТЕРОСТАТ таблетки 20 мг * 30 СТАДА
Библиография
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1853359/
https://en.wikipedia.org/wiki/Familial_hypercholesterolemia
https://emedicine.medscape.com/article/121298-medication
https://en.wikipedia.org/wiki/Hyperlipidemia
Коментари към Чиста хиперхолестеролемия МКБ E78.0