Разстройства на обмяната на пирувата и глюконеогенезата МКБ E74.4
Разстройства на обмяната на пирувата и глюконеогенезата включва недоимък на фосфоенолпируват карбоксикиназа, недоимък на пироват карбоксилаза и недоимък на пироват дехидрогеназа.
Към разстройства на обмяната на пирувата и глюконеогенезата принадлежи недоимък на фосфоенолпируват карбоксикиназата.
Фосфоенолпируват карбоксикиназа представлява ензим, който има съществено значение в метаболитния път на глюконеогенезата. Той превръща оксалацетат в фосфоенолпируват и въглероден диоксид.
По своята същност това е рядко срещано заболяване, което се проява непосредствено след раждането и в дните след него.
Клинично заболяването се характеризира с хипогликемията, сънливост, която се дължи на хипогликемията, забавен растеж и развитие на детето, наличие на метаболитна ацидоза, която се дължи на натрупване на млечна киселина, иктер с хепатомегалия, генерализирана мускулна слабост.
За диагнозата му основно значение има извършването на специализирани изследвания, клиничната картина, анамнезата и лабораторните изследвания.
Диференциална диагноза се прави с всички заболявания, които имат подобна клинична симптоматика.
Лечението е специфично и се осъществява от лекар специалист.
Към разстройства на обмяната на пирувата и глюконеогенезата принадлежи недоимък на пироват карбоксилаза.
Дефицит на пируват карбоксилаза представлява рядко заболяване, което може да доведе до забавяне на развитието в неонаталния период или в периода на ранно детство.
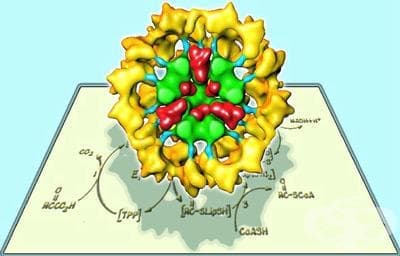
Пируват карбоксилаза е ензим, който играе важна роля в производството на енергия. Той катализира превръщането на пируват до оксалацетат. Оксалоацетат е един от съществените субстрати, необходими за производството цитрат, първия субстрат в глюконеогенезата.
Резултатите от дефицит на пируват карбоксилаза е неизправност на цикъла на лимонената киселина и на глюконеогенезата (както се вижда на схемата по-долу), като по този начин лишава организма с енергия.
Съществуват три типа дефицит на пируват карбоксилаза.
Тип А - характеризира с инфантилност началото, умерено повишение на лактата, глобално забавяне на развитието с умствена изостаналост и оцеляване до зряла възраст.
Тип B - характеризира с начало в неонаталния период, висок лактат и амоняк, нарушение на превръщането лактат до пируват и смърт в рамките на първите няколко месеца от живота.
Type C - най-доброкачествения вариант от трите. Характеризира се с епизоди на лека до умерена лактатна ацидоза, без никакви неврологични или когнитивни симптоми.
За да се потвърди диагнозата е необходима оценка на пациента от специалист в метаболизма и генетичните заболявания. Генетичното консултиране за родители е важно, за да се определи рискът за рецидив бъдещи бременности.
Диференциална диагноза се прави с всички заболявания, които имат подобна клинична симптоматика.
Лечението е специфично и се осъществява от лекар специалист.
Към разстройства на обмяната на пирувата и глюконеогенезата принадлежи недоимък на пироват дехидрогеназа.
Пируват дехидрогеназен дефицит е едно от най-честите невродегенеративни заболявания, свързани с нарушения митохондриална метаболизъм. По своята същност това представлява едно X-свързано заболяване, което показва хетерогенни характеристики.
Клинично заболяването може да има неврологична форма за, която се характеризира с гърчове и / или невропатологичните спазми. Това представяне на заболяването обикновено прогресира до умствено изоставане, микроцефалия, слепота.
Пренатално началото може да бъде представено с неспецифични симптоми като ниска оценка Апгар и раждане на деца, които са малки за гестационната си възраст.
За диагнозата му основно значение има извършването на специализирани изследвания, клиничната картина, анамнезата и лабораторните изследвания.
Диференциална диагноза се прави с всички заболявания, които имат подобна клинична симптоматика.
Лечението е специфично и се осъществява от лекар специалист.
Коментари към Разстройства на обмяната на пирувата и глюконеогенезата МКБ E74.4