Остра промиелоцитна левкемия МКБ C92.4
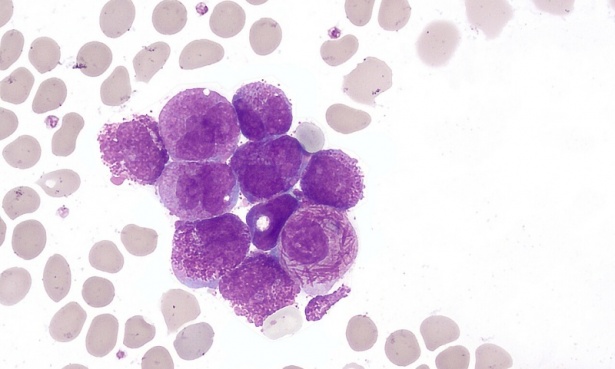
Към миелоидните левкемии се причислява и така наречената остра промиелоцитна левкемия.
Дължи се на арест в диференциацията на левкоцитите в етапа на промиелоцитите. По системата FAB се класифицира като клас М3 левкемия. Асоциира се с транслокация между хромозомите 15 и 17.
Първото описание на това заболяване датира от 1950 г. в Норвегия и Франция като много фатален хеморагичен синдром. През 1959 г. Жан Бернар описва връзката между острата промиелоцитна левкемия (ОПЛ) и тежката хеморагична диатеза, които водят до дисеминирана интраваскуларна коагулация и хиперфибринолиза.
Патохистологично ОПЛ се определя от своите цитогенетични отнасяния. Повече от 95 % от случаите се характеризират с балансирана транслокация между хромозома 17q21 и хромозома 15q22, което води до образуването на патологичния протеин PML-RARA.
PML е промиелоцитен ген, които се кодира от хромозома 15 и се експресира повсеместно, като участва в апоптозата и туморната експресия.
RARA е генът, отговарящ за алфа рецептора на ретиноловата киселина, кодиран от дългото рамо на хромозома 17 и експресиран основно в хематопоетичните клетки. В присъствието на ретинолова киселина, RARA се активира и води до терминалната диференциация на промиелоцитите. При около 40 % от случаите се наблюдават и допълнителни хромозомни аномалии, като тризомия 8 и изохромозома 17.
Този тип миелоидна левкемия съставлява 5-15 % от всички случаи на левкемия при възрастни, като не се наблюдават съществени различия в честотата сред мъжете и жените. Средната възраст на изява е около 40 години.
Различават се няколко морфологични варианта:
- Хипергрануларен подтип (класически М3): наблюдават се множество пръчици на Auer, струпвания от гранулиран материал, съдържащ лизозоми, пероксидаза, лизозомни ензими и големи кристални включвания. Костният мозък обикновено е хиперцелуларен.
- Микрогрануларен подтип: съставлява около 25 % от случаите, като в цитоплазмата се наблюдават фини, тъмни гранули, а пръчиците на Auer са редки.
- Хипербазофилен подтип: налице са малък брой гранули, липсват пръчици на Auer, а ядреноцитоплазменият индекс е повишен.
- PLZF-RAR alfa подтип: наподобява хипергрануларния подтип, с разликата, че тук има по-малко гранули и пръчици на Auer.

Изображение: The Armed Forces Institute of Pathology (AFIP), Public domain, via Wikimedia Commons
Голяма част от клиничната симптоматика се припокрива с тази при остра миелоидна левкемия и включва анемия, слабост, лесна уморяемост, тромбоцитопатия, левкопения, висока телесна температура, чести инфекции и други. При голяма част от пациентите се наблюдава панцитопения.
За разлика от ОМЛ, тук се наблюдава и коагулопатия, описана като дисеминирана интравазална коагулация (ДИК синдром) в съчетание с хиперфибринолиза.
Състоянието протича с ниски нива на плазминоген, инхибитор на алфа2 плазмин и плазминоген активиращ инхибитор 1. Налице е повишена експресия на анексин 2, който е рецептор за плазминогена и плазминоген-активиращия фактор, разположен на повърхността на левкемичните промиелоцити. Това води до свръхпродукция на плазмин и свързаната с това фибринолиза.
При почти половината от нелекуваните пациенти се наблюдават белодробни и мозъчни кръвоизливи, които могат да бъдат фатални.
Чести клинични признаци са множеството петехии и екхимози, кървенето от венците и изразената бледност на пациента. При ангажиране на централната нервна система от процеса са налице и неврологични нарушения и главоболие.
При диференциалната диагноза е необходимо да се имат предвид следните състояния и заболявания:
- левкемоидна реакция към инфекциозни заболявания
- вирусни инфекции, особено инфекциозна мононуклеоза
- недостиг на витамин В12
- остра лимфобластна левкемия
- остра миелоидна левкемия
- апластична анемия
- миелодиспластичен синдром
Диагнозата при остра промиелоцитна левкемия се базира на клиничните прояви, данните от лабораторните и образните изследвания.
Необходимо е извършване на пълна кръвна картина с диференциално броене, натривки от периферна кръв, цялостен метаболитен профил за изходното ниво на показателите, отразяващи бъбречната и чернодробната функция, протромбиново време и активирано парциално тромбопластиново време (аРТТ). С особено висока информативна стойност е провеждането на аспирационна биопсия на костен мозък и последваща цитометрия и цитогенетичен анализ.
Лечението се подразделя на три фази, а именно индуциращо, консолидиращо и поддържащо. Изключително важно средство, прилагано и в трите фази на лечение е транс ретиноловата киселина. При 25-50 % от пациентите, в резултат на терапията, се развива синдром на ретиноловата киселина, който протича с треска, хипотония, респираторен дистрес, хипоксемия и остра бъбречна недостатъчност.
Транс ретиноловата киселина обикновено се прилага в комбинация с даунорубицин, цитарабин, идарубицин или арсенов триоксид. Възможно е и извършване на трансплантация на костен мозък при някои пациенти.
Прогнозата при пациентите с остра промиелоцитна левкемия е относително благоприятна.
Заглавно изображение: Максим Гуцалюк, CC BY-SA 4.0, via Wikimedia Commons
Симптоми и признаци при Остра промиелоцитна левкемия МКБ C92.4
- Главоболие
- Коремна болка
- Умора
- Усещане за отпадналост
- Повишена телесна температура
- Недостиг на въздух (диспнея)
Коментари към Остра промиелоцитна левкемия МКБ C92.4