Синдроми на вродени аномалии със засягане предимно на крайниците МКБ Q87.2
В рубриката синдроми на вродени аномалии със засягане предимно на крайниците са включват:
- синдром на Holt-Oram
- синдром на Klippel-Trénaunay-Weber
- синдром на (липса) недоразвитите на нокти-патела
- синдром на Rubinstein-Taybi
- синдром на sirenomelia
- синдром на тромбоцитопения и липса на лъчева кост (TAR)
- синдром на VATER
Синдромът на Холт-Орам (Holt-Oram) е генетично, автозомно доминантно заболяване, което се характеризира със скелетни аномалии на ръцете и рамената (горните крайници) и сърдечни проблеми.
Наблюдава се присъствието на най-малко една аномалия в костите на китката. Често тези аномалии на китковите кости могат да бъдат открити само чрез рентгеново изследване. Могат да се наблюдават и допълнителни костни аномалии, включително липсващ или анормално дълъг палец, който прилича на пръст, частична или пълна липса на кости в предмишницата, недоразвита кост на горната част на ръката, както и аномалии на костите на плешките. Тези скелетни аномалии могат да засегнат едната или и двете ръце. Аномалиите на костите могат да бъдат еднакви или различни от всяка страна. В случаите, когато скелетните аномалии не са еднакви от двете страни на тялото, лявата страна е обикновено по-силно засегната от дясната страна.
Около 75 процента от хората със синдром на Холт-Орам със сърдечни проблеми, които могат да бъдат животозастрашаващи. Най-честият проблем е дефект в мускулната стена (преграда), която разделя лявата и дясната страна на сърцето. Другите сърдечни проблеми, които могат да бъдат наблюдавани са предсърдно-септален дефект (ASD) и камерен преграден дефект (VSD). Някои хора имат и сърдечно заболяване, което се изразява в нарушена проводимост, причинена от аномалии в електрическата система. Сърдечната електрическа система е жизненоважна, тъй като тя координира контракциите на сърдечните камери. Нарушенията в сърдечната проводимост могат да доведат до проблеми, като по-бавен от нормалния сърдечен ритъм (брадикардия) или по-бърз (тахикардия) и некоординирано свиване на сърдечния мускул (мъждене). Нарушенията в сърдечната проводимост могат да се появят заедно с други дефекти на сърцето (като ASD или VSD) или като единственият проблем със сърцето при хора със синдром на Холт-Орам.
Причината за синдрома на Холт-Орам са множество мутации в гена TBX5. Намира се в дългото рамо (Q) на хромозома 12 (12q24.1). Този ген осигурява инструкции за получаване на протеин, който играе роля в развитието на сърцето и горните крайници преди раждането. По-специално, този ген е важен за осъществяването на развитието на сърдечните камери. Генът TBX5 също така играе важна роля и в регулирането на развитието на костите на ръката и китката. Мутациите в този ген вероятно нарушават развитието на сърдечни камери и горните крайници, което води до характерните черти на синдром на Холт-Орам.
Диагнозата синдром на Холт-Орам се основава на физически констатации и фамилна анамнеза. Рентгеново изследване се извършва на горните крайници за евентуално откриване на малформации. Ехокардиография, MRI и електрокардиографията се използват за определяне на наличието и тежестта на дефекти на сърцето и / или нарушения в сърдечната проводимост. Молекулярно генетично изследване за ген TBX5 е достоверно за потвърждаване на диагнозата. Някои индивиди с леки симптоми не са диагностицирани. Пренаталното ултразвуково изследване на сърцето на плода (ехокардиография) се използва, за да се диагностицират сърдечни малформации при деца на засегната майка.
Лечението на синдрома на Холт-Орам е насочено към управление на специфичните симптоми, които са индивидуални за всеки. Специфичните терапии за синдрома на Холт-Орам са симптоматични и поддържащи.
Синдромът на Klippel-Trénaunay-Weber (KTS) е състояние, което засяга развитието на кръвоносните съдове, меките тъкани (като кожата и мускулите), както и костите. Този синдром също спада към синдроми на вродени аномалии със засягане предимно на крайниците.
Има три характерни черти за този синдром:
- червен белег по рождение
- ненормален свръхрастеж на меките тъкани и костите
- венозни малформации
Синдромът на Klippel-Trenaunay се дължи на мутации в гена PIK3CA. Този ген осигурява инструкции за синтез на протеин р110 алфа (p110α), който е част (субединица) от ензим, наречен фосфатидилинозитол 3-киназа (PI3K). PI3K играе роля в химически сигнализация, която е важна за много клетъчни дейности, включително клетъчен растеж и делене (пролиферация), движение (миграция) на клетки и клетъчното оцеляване. Тези функции на PI3K са важни за развитието на тъканите в тялото.
Генните мутации в PIK3CA променят p110α протеина. Променената субединица прави PI3K необичайно активен, което позволява на клетките да растат и да се делят непрекъснато. Повишената клетъчна пролиферация води до анормален растеж на костите, меките тъкани и кръвоносните съдове. Тъй като не всички със синдром на Klippel-Trenaunay имат мутация в гена PIK3CA, възможно е мутации в неидентифицирани гени да предизвикат това състояние.
Повечето хора се раждат направо с характерните червени петна. Този тип петна се причиняват от подуване на малките кръвоносни съдове в близост до повърхността на кожата. Петната обикновено са плоски и могат да варират от бледо розово до наситено кафяви на цвят, което обикновено обхваща част от единия крайник. Засегнатата област може да стане по-светла или по-тъмна с възрастта. От време на време, в петната се развиват малки червени мехури, които спонтанно могат да се отворят на повърхността и да изкървят.
Прекомерният растеж на костите и меките тъкани започват още в ранна детска възраст. Обикновено този анормален растеж е ограничен до един крайник, даже най-често един крак. Въпреки това, свръхрастежът може да повлияе ръцете или, по-рядко, торса. Неправилното разрастване може да причини болка, чувство на тежест и намалено движение в засегнатата зона. Ако свръхрастежът се наблюдава само в единия крак, то той може да е по-дълъг от другия и да създава проблеми с ходенето.
Малформациите на вените са третата голяма черта на синдрома на Klippel-Trenaunay. Тези аномалии се изразяват в разширени вени, които са подути и усукани вени в близост до повърхността на кожата и много често причиняват болка. Разширените вени обикновено се появяват по стените на горните крайници и прасците. Разширените вени могат да бъдат и дълбоките вени на крайниците, но това е необичайно за синдрома. Малформациите на дълбоките вени увеличават риска от дълбока венозна тромбоза на (DVT). Ако DVT пътува през кръвния поток и навлезе в белодробното кръвообращение, белодробна емболия (PE). Белодробната емболия е спешно състояние, което може да доведе до фатален край в рамките на няколко минути.
Други усложнения на синдрома на Klippel-Trenaunay могат да бъдат инфекции на кожата (целулит), оток, причинен от натрупване на течност (лимфедем), и вътрешно кървене от анормални кръвоносни съдове. По-рядко синдактилия или полидактилия.
Диагностиката се извършва чрез ултразвуково изследване, ангиография, ядрено-магнитен резонанс (ЯМР), Компютърна томография (КТ), венография, сцинтиграфия, лимфография, рентгенография.
Лечението е симптоматично и поддържащо.
Синдромът на (липса) недоразвитите на нокти-патела се характеризира с нарушения на ноктите, коленете, лактите, и таза. Характеристиките варират по тежест между засегнатите лица, дори и сред членовете на едно семейство. Спада към синдроми на вродени аномалии със засягане предимно на крайниците.
Причините за синдрома са множество генни мутации на гена LMX1B. Той осигурява инструкции за производство на протеин, който се свързва със специфични региони на ДНК и регулира активността на други гени. Въз основа на тази роля на LMX1B протеинът се нарича фактор на транскрипция. Протеинът LMX1B е особено важен по време на ранното ембрионално развитие на крайниците, бъбреците и очите.
Мутации в LMX1B гена водят до производството на необичайно малък, нефункционален протеин или със засягане способността на протеина да се свързва с ДНК. Не е ясно как мутации в LMX1B гена предизвикват признаците и симптомите на синдрома (липса) недоразвитите на нокти-патела.
Унаследява се по автозомно доминантен път.
Аномалии на ноктите се наблюдават в почти всички случаи, като те могат да отсъстват или да са недоразвити и обезцветени, набраздени или други. Ноктите на ръцете се засягат по-често от ноктите на краката. Обикновено са най-тежко засегнати. В много от хората областта, в основата на ноктите е триъгълна, вместо обичайната форма на полумесец. Обикновено нокътните изменения са симетрични.
Много често се откриват и скелетни аномалии, които обхващат коленете, лактите и бедрата. Капачките на коленете са малки с неправилна форма или отсъстват напълно, дислокацията на пателата е обща. Някои хора в резултат може да не са в състояние напълно да си отворят ръцете или да си обръща дланите нагоре, като същевременно поддържат лактите. Лактите могат да бъдат под ъгъл навън (кубитус валгус). Понякога се откриват израстъци на илиачните кости на таза (илиачни рога). Тези промени се откриват само при рентгеново изследване. Илиачните рога са много чести при хора със синдром на липса или недоразвитите на ноктите-пателата и се срещат рядко при хора, които не страдат от този синдром.
Другите части, които могат да бъдат засегнати са най-вече очите и бъбреците. Хората с това заболяване са изложени на риск от развитие на повишено вътреочно налягане, в резултат развитие на глаукома с отворен ъгъл още в много ранна възраст. Може да се отключи и бъбречно заболяване, което по-късно да прогресира до бъбречна недостатъчност.
Диагнозата се поставя на базата на наличните клинични данни.
Лечението е симптоматично и поддържащо.
Синдромът на Rubinstein-Taybi е състояние, характеризиращо се с нисък ръст, умерена до тежка умствена изостаналост, отличителни черти на лицето и широки палци на краката.
Допълнителни характеристики на заболяването могат да включват очни аномалии, сърдечни и бъбречни дефекти, проблеми със зъбите и затлъстяване. Тези признаци и симптоми варират между засегнатите индивиди.
Хората с това заболяване имат повишен риск от развитие на тумори, включително и някои видове мозъчни тумори. Ракът на кръвообразуващата тъкан (левкемия) се развива по-често при хората със синдром на Rubinstein-Taybi, спрямо здравите хора.
Унаследява се автозомно доминантно. Въпреки това повечето случаи са резултат от нови мутации в гена (de novo) и се срещат при хора, без история на заболяването в семейството им.
Мутации в гена CREBBP са отговорни за някои от случаите на синдрома на Rubinstein-Taybi. Генът CREBBP осигурява инструкции за получаване на протеин, който помага за контролирането на активността на много други гени. Този протеин, играе важна роля в регулирането на клетъчния растеж и делене и е от съществено значение за нормалното развитие на плода. Ако едно копие на гена на CREBBP се заличава или е мутирал, клетки синтезират само половината от нормалното количество на CREB-свързващ протеин. Въпреки намаляването на размера, този протеин нарушава нормалното развитие преди и след раждане. Мутации в гена EP300 предизвиква малък процент от случаите на синдрома на Rubinstein-Taybi.
Признаците и симптомите на това заболяване при хора с мутации в EP300 гена са подобни на тези с мутации в гена на CREBBP. Въпреки това, проучвания показват, че мутации в EP300 гена могат да бъдат свързани с леки скелетни промени в ръцете и краката.
Някои случаи на тежък синдром на Rubinstein-Taybi са причинени от делеция на късото рамо на 16 хромозома. Няколко гени, включително ген CREBBP, отпадат в резултат от делецията. Смята се, че загубата на множество гени в тази област вероятно водят до сериозни усложнения, свързани с тежък синдром на Rubinstein-Taybi. Въпреки това около половината от хората със синдром на Rubinstein-Taybi не разполагат с установена мутация в гена CREBBP или EP300 или делеция в хромозома 16.
Основните характеристики на синдрома са:
- изоставане на психофизическото развитие
- лицева дисплазия
- аномалии на пръстите на ръцете
- по-нисък среден растеж
- ниска костна възраст
- умствена изостаналост (100%), като в повечето случаи е тежка
- забавено моторно и забавено развитие на речта, което се изразява в прекомерна разсеяност, лоша концентрация, затруднения в говора придобиване
- сложна краниофациална дисплазия включва брахицефалиа, микроцефалия, късно затваряне на голямата фонтанела, бавен растеж на косата, високо разположени и извити вежди, широк нос, епикантус, дълги мигли, птоза, широк корен на носа, наведен нос (клюнест нос), хипоплазия на нос, умерена ретрогнатия, гримаса, наподобяваща усмивка, дъговидно високо разположено небце
- от страна на очите са маркирани страбизъм и нарушена рефракция (пречупване) на окото
- предсърдни деформации
- аномалии на пръстите са полидактилия, скъсяване, Hallux валгус на интерфалангеалните стави, удвояване на нокътната фаланга на първите пръстите
- лордоза, кифоза, сколиоза, аномалии на гръдната кост и ребрата
- сплескани тазови кости
Вполовината от случаите се появят хирзутизъм, ярко червен вал върху кожата на челото, шията
малформациите на вътрешните органи включват персистиращ дуктус артериозус и сърдечни пороци, едностранна бъбречна аплазия, удвояване на бъбреците, хидронефроза, разширяване на уретерите или стеноза, дивертикул на пикочния мехур, крипторхизъм, нарушен строеж на белодробния паренхим, агенезия или аплазия на мазолестото тяло.
Рядко, синдромът на Rubinstein-Taybi причинява сериозни усложнения, като забавено наддаване на тегло и нарушения в растежа, в сравнение с очакваната за възрастта норма (невъзможност да се развиват) и животозастрашаващи инфекции. Бебета, родени с тежката форма на заболяването обикновено оцеляват само в ранна детска възраст.
Диагнозата се базира на данните от клиничните и генетичните изследвания.
Лечението е симптоматично и поддържащо.
Синдромът на сиреномиелия представлява състояние, при което се наблюдава срастване на долните крайници. Нарича се още синдромът на русалката. Този синдром също спада към синдроми на вродени аномалии със засягане предимно на крайниците.
Това е изключително рядко вродено заболяване. Засегнатите бебета се раждат с частично или напълно слети крака. Допълнителни малформации в различни системи като пикочно-полова, храносмилателна, аномалии в лумбосакралната част на гръбначния стълб и таза, липса или недостатъчно развитите на един или и двата бъбрека.
Засегнатите деца могат да имат един крак или липса на двата крака. Опашната кост обикновено отсъства и сакрума отсъства частично или напълно. Могат да се наблюдават също така неперфориран анус, спина бифида и сърдечни малформации.
Сиреномелия е често фатално състояние по време на неонаталния период. В повечето случаи синдромът се развива случайно, без видима причина (спорадично).
Сиреномелията се свързва с тежки животозастрашаващи усложнения и често е фатално в първите години от живота. Въпреки това, оцеляване извън ранна детска възраст в по-късно детство или млада възраст е докладвано в шепа случаи.
Характерната констатация на сиреномиелия е частично или пълно сливане на долната част на краката. Степента на тежест е силно променлива. Засегнатите деца могат да имат само една бедрена кост (фемур) или могат да имат две бедрени кости. Засегнатите деца могат да имат един крак или да бъдат без крака, или да имат два крака, които могат да се въртят така, че задната част на крака е обърната напред.
Наблюдават се също така различни урогенитални аномалии, включително липса на един или и на двата бъбрека (бъбречна агенезия), кистозна малформация на бъбреците, отсъстващ пикочен мехур, стесняване на уретрата (уретрална атрезия). В допълнение, могат да имат неперфориран анус, състояние, при което е блокиран аналния отвор или канал, който обикновено се свързва с ануса и най-ниската част на дебелото черво (ректум).
Бебета със сиреномиелия също могат да имат аномалии, засягащи сакралните и лумбалните прешлени. При някои пациенти може да се наблюдава изкривяване на гръбначния стълб напред (лордоза), липса на външни полови органи, липса на вътрешни органи като далака, жлъчния мехур, дефекти на коремната стена (омфалоцеле), менингомиелоцеле, сърдечни аномалии, белодробна хипоплазия (недоразвитие).
Точната причина за сиреномелия е неизвестна. Смята се, че факторите на околната среда и генетични фактори играят роля в развитието на заболяването. В повечето случаи възникват без видима причина (спорадично).
Различни генетични фактори могат да допринесат за това разстройство у различни хора (генетична хетерогенност). Смята се също така, че нарушения в ранното развитие на съдовата система могат да бъдат причина. Понякога се открива една единствена артерия, разклоняваща се от коремната аорта, вместо обичайните две. Артериите в опашната област са недоразвити. Така притокът на кръв към опашната кост и долните крайници става невъзможен или отслабен, в резултат на което те не се развиват или развитието им остава в един непълен етап.
Диагнозата може да се постави пренатално чрез ултразвукова диагностика по време на второто тримесечие.
Лечението е симптоматично и поддържащо.
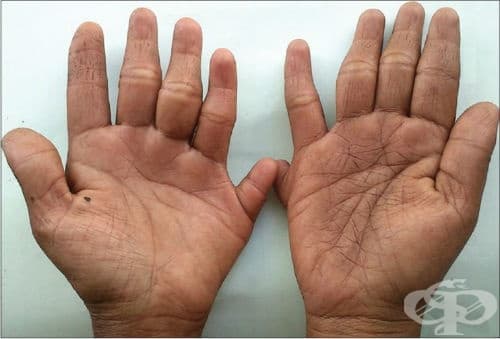
Синдромът на тромбоцитопения с липса на лъчева кост (TAR) се характеризира с дефицит на кръвни клетки, участващи в кръвосъсирването (тромбоцити). Този недостиг се означава с термина тромбоцитопения и обикновено се появява по време на ранна детска възраст и се стабилизира частично или напълно с течението на времето. Спада към синдроми на вродени аномалии със засягане предимно на крайниците.
Тромбоцитопения предотвратява нормалното съсирване на кръвта, което води до лесно нараняване и чести кръвотечения от носа. Потенциално животозастрашаващи случаи на тежко кървене (кръвоизлив) може да се появят в мозъка и други органи, особено по време на първата година от живота. Кръвоизливите могат да увредят мозъка и да доведат до трайно умствено увреждане. Засегнатите деца, които преживяват този период, обикновено имат нормална продължителност на живота и нормално интелектуално развитие.
Синдромът на TAR се различава от другите синдроми, протичащи с липса на радиус по това, че палецът не отсъства. При другите синдроми, характеризиращи се с липса на радиус обикновено липсва и палецът.
Други характерни симптоми са нисък ръст, допълнителни скелетни аномалии, включително недоразвитост на други кости на ръцете и краката. Засегнатите лица могат да имат и малформации на сърцето и бъбреците, необичайни черти на лицето, включително малка долна челюст (микрогнатия), издадено напред чело и ниско поставени уши. Около половината от засегнатите лица са алергични към краве мляко, което може допълнително да влоши тромбоцитопенията, свързана с това заболяване.
Като причина за заболяването се смятат множество мутаци в гена RBM8A, който осигурява инструкции за получаване на протеин, наречен РНК-свързващ протеин 8А. Този протеин се смята, че участва в няколко важни клетъчни функции, свързани с производството на други протеини.
Синдромът се унаследява автозомно рецесивно.
Лечението е свързано с многократни инфузии на тромбоцитна маса, симптоматично и поддържащо. Критичният период е първата година и понякога втората година от живота. За повечето хора с TAR, броят на тромбоцитите се подобрява, тъй като те растат от детството.
Синдромът на VATER е набор от вродени дефекти, които често се срещат заедно. Инициалите във V.A.T.E.R. синдром се отнасят до пет различни области, в които може да има отклонения:
- дефекти в прешлени
- анална атрезия
- трахео-езофагеала фистула
- бъбречни аномалии
Може да има и сърдечни дефекти и дефекти в крайниците, които променят акронима във V.A.C.T.E.R.L. Засегнатото дет не е задължително да има проблем във всяка една от посочените области, но има съзвездие от вродени дефекти, включващи много от областите.
Дефектите в прешлените могат да включват разнообразни деформации, разтопени прешлени, липса на прешлен или наличие на допълнителен. При част от хората тези деформации изискват хирургична намеса, тъй като ограничават ежедневието на болния или причиняват здравословни проблеми.
Аналната атрезия от своя страна може да бъде придружена от аномалии на половите органи и пикочните пътища.
Сърдечните дефекти могат да варират по тежест от животозастрашаващи до фин дефект, който не предизвиква здравословни проблеми.
Много често при тези болни се среща и трахео-езофагеална фистула, която представлява ненормална връзка (фистула) между хранопровода и трахеята. Трахео-езофагеална фистула може да доведе до проблеми с дишането и храненето в началото на живота и обикновено изисква хирургична корекция в ранна детска възраст.
Засегнатите лица могат да имат липса на един или двата бъбрека или са ненормално развити, безформени, които могат да повлияят бъбречната функция.
Аномалиите на крайниците най-често включват слабо развити или липсващи палци или недоразвити предмишници и китки.
Важно за синдрома е, че не повлиява интелектуалното развитите.
Диагнозата е изключително клинична.
Лечението е симптоматично и поддържащо.
Коментари към Синдроми на вродени аномалии със засягане предимно на крайниците МКБ Q87.2