Ограничена атрофия на главния мозък МКБ G31.0
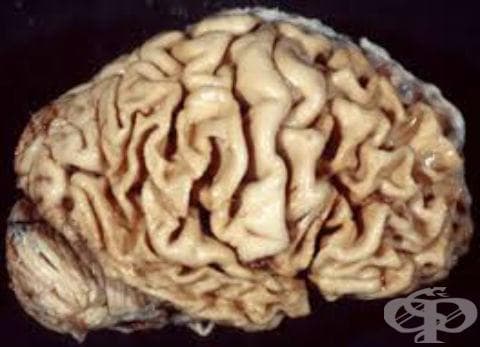
В рубриката ограничена атрофия на главния мозък са разгледани:
- болест на Pick
- прогресираща изолирана афазия
БОЛЕСТ НА ПИК (Pick)
Фронтотемпоралната деменция, наричана още фронтотемпорална дегенеративна болест или фронтотемпорално неврокогнитивно разстройство, обхваща няколко вида деменция, включваща прогресивна дегенерация на фронталните и темпоралните дялове на мозъка.
Това е вторият най-разпространен тип деменция с ранно начало след болестта на Алцхаймер, като обикновено се представя като поведенческо или езиково разстройство с постепенно начало.
Характеристиките на заболяването са описани за първи път от Арнолд Пик между 1892 и 1906 г., като името "болест на Пик" е въведено през 1922 година.
Болестта на Pick se представя с една от двете добре разпознати фенотипни форми на когнитивна дисфункция: поведенчески вариант на фронтотемпорална деменция и семантична вариант на първична прогресивна афазия.
История
Характеристиките на фронтотемпоралната деменция са описани за първи път от чешкия психиатър Arnold Pick между 1892 и 1906 година. Името болест на Пик е въведено през 1922 година, като този термин сега е запазен само за поведенческия вариант на заболяването, който показва наличието на характерните тела на Пик и клетки на Пик, които са описани за първи път от Алоис Алцхаймер през 1911 г.
През 1989 година Snowden предлага термина семантична деменция, за да опише пациент с преобладаваща лява темпорална атрофия и афазия, описани от Pick. Първите критерии за фронтотемпорална деменция са разработени през 1994 г. Клиничните диагностични критерии са ревизирани в края на 1990 г., когато спектърът на заболяването се разделя на поведенчески вариант, вариант на нефлуентна афазия и вариант на семантична деменция. Последната ревизия на критериите за клинични изследвания е от Международния консорциум за критериите за поведенчески варианти през 2011 година.
Епидемиология
Фамилните форми на болестта на Пик могат да се наблюдават по-често в Европа (особено в скандинавските страни). Приблизителната честота варира от 7 до 43 случая на 100 000 души от населението.
Фамилните форми на деменции при болест на Pick, свързани с хромозома 17, могат да бъдат особено чести при хора от скандинавските страни, като може да съставляват до 17% от деменциите в тази популация.
Болестта на Пик се среща в по-млада възрастова група в сравнение с деменцията от типа на Алцхаймер, като пиковата честота се среща при лица на възраст 55-65 години.
Случаите са склонни да имат променливо начало като начало е докладвано още през второто десетилетие и най-късно през десетото десетилетие. Около 40% от пациентите имат положителна фамилна анамнеза за деменция, което е значително по-високо от повечето други невродегенеративни заболявания.
Етиология
Болестта на Пик е таупатия. Таупатиите са синдроми, които възникват вторично в резултат на отлагането на анормални форми на тау протеин в мозъка.
Тау протеинът е силно разтворим протеин, свързан с микротубули (MAPT) и насърчава полимеризацията и стабилизирането на микротубулите. Тау протеинът в мозъка е хетерогенен поради алтернативни форми на сплайсинг и пост-транслационни модификации.
Тау протеинът при болестта на Пик има уникални характеристики. Състои се от остатъци K254-F378 на 3R tau, докато други таупатии (включително болестта на Алцхаймер, прогресивна супрануклеарна парализа и кортикобазална ганглийна дегенерация) имат или 4Rtau, или комбинация от 3R и 4Rtau.
Натрупването на анормален протеин води до прогресивна невронална дисфункция и загуба на мозъчно вещество.
Класификация
Различните таупатии могат да бъдат диференцирани въз основа на предилекционните области на засягане на мозъка и/или засягане на специфични клетки/клетъчни групи.
Фронтотемпоралните подтипове включват:
- болест на Пик (Picks disease)
- кортикобазална дегенерация (corticobasal degeneration, CBD)
- прогресивна супрануклеарна парализа (progressive supranuclear palsy, PSP)
- глобуларна глиална тауопатия (globular glial tauopathy, GGT)
- първична свързана с възрастта тауопатия (primary age-related tauopathy, PART)
Всеки подтип е относително рядък и представлява синдром с ранно начало, свързани с фронтотемпорална лобарна дегенерация, която се характеризира с прогресивна загуба на неврони, засягащи предимно фронталните или темпоралните лобове, и типична загуба на повече от 70% от вретеновидни неврони, докато други видове неврони остават непокътнати.
Трите основни подтипа или вариантни синдрома са поведенчески вариант и два варианта на първична прогресивна афазия: семантична и нефлуентна.
Поведенческият вариант на фронтотемпорална деменция е бил известен преди като болест на Pick и е най-често срещаният тип. Поведението може да се промени по един от двата начина - може да се промени до импулсивно, действайки по социално неприемливи начини, или може да се промени до апатичен вид. Около 12-13% от хората с този вариант развиват заболяване на моторните неврони.
Семантичната деменция се характеризира със загуба на семантично разбиране, което води до нарушено разбиране на думите. Речта обаче остава плавна и граматически правилна.
Прогресивната нефлуентна афазия (PNFA) се характеризира с прогресивни затруднения на речта.
Патоморфология
Има три основни хистологични подтипа: FTLD-tau, FTLD-TDP и FTLD-FUS. В редки случаи при пациенти с клинична фронтотемпоралната деменция е установено, че имат промени, съответстващи на болестта на Алцхаймер при аутопсия. Най-тежката мозъчна атрофия изглежда е свързана с поведенчески вариант на заболяването и кортикобазална дегенерация.
По отношение на генетичните дефекти, които са открити, повторната експанзия в гена C9orf72 се счита за основен принос към фронтотемпорална лобарна дегенерация, въпреки че дефектите в гените GRN и MAPT също са свързани с нарушенията.
Увреждането на ДНК и дефектното възстановяване на такива увреждания са етиологично свързани с различни невродегенеративни заболявания, включително фронтотемпоралната деменция.
Клинична картина
Фронтотемпоралната лобарна дегенерация е невропатологично наименование, което обхваща група от невродегенеративни заболявания на фронталните и предните темпорални лобове, свързани със специфични патологии, включително патология на Pick.
Първичното увреждане на когнитивните способности не включва нарушение на съзнанието или разсеяност. Началото на поведенческа и когнитивна дисфункция при индивиди с болестта на Pick може да бъде трудно за идентифициране. В много случаи индивидите с болестта на Пик се представят добре при бързи когнитивни скринингови тестове.
Най-често срещаният клиничен подтип е поведенческият вариант на фронтотемпоралната деменция, който съставлява приблизително повече от половината случаи. Заболяването се характеризира с ранни и често тежко инвалидизиращи промени в личността и поведението, които могат да бъдат предшествани от дълга фаза на субклинични поведенчески промени и социални смущения, често възникващи при липса на когнитивно увреждане.
Установяват се промени във вниманието, поведенческата функция и когнитивни нарушения при пресимптоматични пациенти.
Дисоциацията от семейството, компулсивно разстройство при пазаруване (ониомания), вулгарни речеви характеристики, крещене, неспособност за контролиране на емоциите, поведението, личността и темперамента са характерни модели на социално поведение.
Пациентите с орбитофронтална дисфункция могат да станат прекалено приятелски настроени и може да изглеждат неуместно откровени в разговорите си. Те стават по-импулсивни, склонни към пристрастяване, раздразнителни и е вероятно да проявят антисоциално поведение.
Може да се отбележи обсесивно-компулсивно или повтарящо се стереотипно поведение.
Пациентите с дорзомедиална или дорзолатерална фронтална дисфункция могат да демонстрират липса на безпокойство, апатия или намалена спонтанност, което може да прогресира до мутизъм и неподвижност в крайните стадии на заболяването.
Пациенти със загуба на сиво вещество в двустранния постеролатерален орбитофронтален кортекс може да демонстрират промени в хранителното поведение, като патологично пристрастяване към сладкото. Пациентите с по-фокална загуба на сиво вещество в антеролатералния орбитофронтален кортекс могат да развият хиперфагия.
Когнитивните симптоми са свързани с лоша преценка, невнимание, разсеяност, загуба на способност за планиране и дезорганизация.
Може да възникне инконтиненция, която е с тенденция да се появява по-рано в сравнение с болестта на Алцхаймер.
Може да възникне паркинсонизъм със съпътстващите прояви на ригидност и нарушение на походката. Тежкият или ранен паркинсонизъм показва алтернативна диагноза, като кортикобазална ганглийна дегенерация, дифузна болест с телца на Lewy или прогресивна супрануклеарна парализа.
Първична прогресивна афазия
Първото систематично описание на случай на тази форма е публикувано през 1982 г. от Marsel Mesulam.
Възрастта на поява варира, като най-често е между 55 и 70 години.
В семантичния вариант се установява преференциално участие на речта и семантичен анализ.
Забелязват се дефицити в назоваването на една дума и в разбирането. Това има тенденция да засяга елементи с ниска честота (по-малко познати) в ранните етапи. Семантичната парафазия е честа. Може да се забележи аномия.
С прогресията на заболяването разпознаването на обекти е нарушено и това засяга всички сензорни модалности (например зрителна агнозия). Отбелязва се повърхностна дислексия и повърхностна дисграфия.
Някои пациенти може да имат затруднения при разпознаването на лицето на човек.
В средно-късните фази промените в поведението могат да бъдат забележими. Те включват дезинхибиране, раздразнителност, промени във вкуса на храната, липса на емпатия и ментални нарушения. Често се наблюдава компулсивно поведение и пациентите могат да покажат прекомерна загриженост.
Увреждането на паметта е сравнително по-леко и епизодичната памет обикновено се запазва по-добре.
Диагноза
Заболяването трудно се диагностицира поради разнообразния характер на свързаните симптоми.
Както при всяка деменция, първоначалната оценка включва изследване на нивата на витамин B-12 и фолиева киселина, изследвания на функцията на щитовидната жлеза, антинуклеарни антитела и изследване на трепонемни антитела за сифилис.
По-нататъшните изследвания включват цереброспинална течност (ликвор) (за хроничен менингит или повишено налягане) и серология на HIV. Тъй като болестта на Алцхаймер е почти винаги в диференциалната диагноза, може да изследва ликвор за Abeta1-42.
Ако се наблюдава изразено нарушение във вниманието, пациентът може да има токсична и/или метаболитна енцефалопатия, а не истинска деменция. При такива пациенти, а също и при пациенти с бърза поява или прогресия на когнитивни оплаквания, е необходимо да се направи токсикологичен скрининг на урината, биохимия, пълна кръвна картина (ПКК) и диференциално броене, чернодробни функционални тестове, ниво на амоняк и скорост на утаяване на еритроцитите. Изследване на злокачествено заболяване, електроенцефалограма (ЕЕГ) и серология на Лаймска болест могат да бъдат показани в избрани случаи.
Ако пациентът има паркинсонизъм или двигателно разстройство, може да се добавят следните изследвания:
- церулоплазмин и изследване на мед в кръв и урина за диагноза на болестта на Wilson
- изследване на периферна кръв за акантоцити
- генетично изследване
Всички пациенти, за които се подозира, че имат болест на Pick, изискват подробна когнитивна оценка.
Образни изследвания
Компютърна томография и ЯМР на мозъка
Ако изследването с ядрено-магнитен резонанс е противопоказано при пациента (например при пейсмейкър или метални импланти), необходимо е да се назначи КТ.
Обикновено семантичното увреждане е свързано с по-голяма левостранна предна темпорална атрофия.
Първичната прогресивна афазия се свързва не само с промени в сивото вещество, но и със значителна дегенерация на бялото вещество.
Лумбална пункция
Лумбална пункция (изследване на ликвора) се извършва при пациенти с атипични характеристики или бързо прогресиращо когнитивно увреждане. Цереброспиналната течност обикновено се изследва за брой клетки, протеин, криптококов антиген, киселинно-устойчиви бактерии, култури, VDRL и цитология, ако клиничната ситуация изисква такова изследване.
Повишени нива на фосфорилиран тау протеин и ниски нива на Abeta1-42 се откриват в ликвор на пациенти с болест на Алцхаймер.
Маркери за болест на Кройцфелд-Якоб, болест на Уипъл на централната нервна система (ЦНС), прогресивна мултифокална левкоенцефалопатия и херпесен енцефалит също могат да бъдат изследвани в гръбначномозъчната течност. Индикацията за такива тестове се определя от клиничната картина и диференциалните диагнози, които се вземат предвид за отделния пациент.
Биопсия на мозъка
Мозъчна биопсия може да се обмисли при изключителни обстоятелства, ако диагнозата е съмнителна и изборът на лечение зависи от резултатите от изследванията. Обикновено това се случва при установяване на атипични характеристики или много бърза прогресия, при която се налага изключване на лечими патологии.
Понякога маркерите на гръбначномозъчната течност могат да избегнат необходимостта от мозъчна биопсия, дори при тези пациенти.
Хистопатологични характеристики
Болестта на Пик е морфологично различна от другите подтипове тау-патии, като хистопатологичните находки включват:
- телца на Пик (Pick)
- изразена загуба на неврони и кортикална атрофия, особено в дясната вентрална и дорзалната фронтална и предна темпорална област.
- разклонени астроцити, характеризиращи се с тау-имунопозитивни отлагания в проксималните астроцитни клонове
Патологията на заболяването на Pick включва:
- Фаза I: лимбично и фронтотемпорално засягане
- Фаза II: засягане на субкортикални структури, включително базални ганглии, таламус и мозъчен ствол
- Фаза III: засягане на първичната моторна кора и предцеребеларното ядро
- Фаза IV: дифузни промени, включително първичната зрителна кора
Лечение
Към момента не съществува лечение на това заболяване.
Целта на терапията е облекчаване на симптомите
Индивидуалните планове за терапия, включително нефармакологични и фармакологични интервенции, са най-подходящи за пациенти с болестта на Pick.
Нефармакологично лечение
Може да включва ограничения за шофиране, осигуряване на безопасна среда и адаптиране, болногледачи и консултации.
Упражнения и физикална терапия - те са полезни особено за пациенти със съпътстващи проблеми с движенията. Такива интервенции могат също да подобрят настроението.
Диета - храните с високо съдържание на захар може да се наложи да бъдат ограничени при някои пациенти. Някои пациенти може да изискват специални грижи, за да предотвратят поставянето на негодни за консумация предмети в устата.
Речева терапия - помага на пациентите да се справят с говорния си дефицит.
Фармакологично лечение
Тъй като при болестта на Pick не се наблюдава дисфункция на кортикалните холинергични системи, употребата на ацетилтрансферазни инхибитори при това състояние не е показана. Инхибиторите на холинестеразата могат дори да влошат поведенческите смущения при тези пациенти.
Проучвания показват нарушение на серотонинергичната и допаминергичната мрежа. Следователно селективните инхибитори на обратното захващане на серотонин (SSRI) (флуоксетин, сертралин, пароксетин, флувоксамин, циталопрам) са ефективни при подпомагане на различни симптоми (дезинхибиране, импулсивност, хранителни разстройства).
При тежки невроповеденчески симптоми, при които SSRI не са полезни, може да се обмисли внимателна и ограничена употреба на антипсихотични лекарства (за предпочитане кветиапин или оланзапин), като се следи внимателно за екстрапирамидни странични ефекти.
Извънболнична помощ
Периодичните последващи грижи са показани за справяне с проблемното поведение или клиничните проблеми на пациента или за преоценка на диагнозата, ако има съмнения.
Ако се появят параноя, депресия или други поведенчески проблеми, фармакологичните лечения могат да бъдат пригодени за справяне с тези проблеми.
Хирургични грижи
Понастоящем не е доказано, че хирургичните лечения са от полза за хора с болест на Пик.
Протичане
Болестта на Пик протича по-кратко от болестта на Алцхаймер, средно около 6 години. При някои лица, чиито основни симптоми са нарушение на речта (първично прогресивна афазия), клиничното протичане може да бъде бавно. В една малка серия тези пациенти са оцелели средно 5 години повече от пациентите с поведенчески симптоми (поведенчески вариант). Пациент с първично прогресивна афазия може да запази способността си да е функционално активен за 10 или повече години след началото.
Прогноза
Подобно на повечето деменции, болестта на Пик бавно прогресира, което води до повишена професионална и лична нетрудоспособност. Въпреки това, малък брой хора, които имат поведенчески смущения, съответстващи на поведенческия вариант на фронтотемпоралната деменция, може да не прогресират - вариант на фенокопия. В предишни проучвания бавна прогресия или никаква прогресия се наблюдава само при пациенти с нелингвистични симптоми - не е наблюдавана първична прогресивна афазия.
Някои пациенти могат да прогресират бавно за изключително дълги периоди. При някои може да се развият артистични или други таланти по време на тяхната деменция - феномен, който може да е свързан с дезинхибиране на "творчески" области на мозъка. Музикалните или артистични вкусове също могат да се променят (например пациентът може да развие внезапен интерес към музика, предназначена за много по-млади слушатели).
Някои пациенти може да са в състояние да придобият нови знания или умения, като например използването на компютърно подпомагана комуникационна система. Това може да бъде полезно при прилагането на техники за поведенческо обучение за оптимизиране на социалната и ежедневната дейност.
Пациентите с това заболяване обикновено оцеляват 7-8 години след началото на заболяването, но преживяемостта може да варира от 2 до 15 години.
ПЪРВИЧНА ПРОГРЕСИВНА АФАЗИЯ
Афазията е неспособност за разбиране, формулиране или вокализиране на говорим или писмен език. Първично прогресивната афазия (ППА) се отнася до групата от невродегенеративни заболявания, показващи постепенно увреждане на говора и езика като първичен симптом без значими когнитивни, физически или поведенчески компоненти.
Класическата афазия обикновено възниква внезапно, както се наблюдава при инсулт. Обратно - ППА се развива постепенно, нарушавайки езиковите функции с течение на времето. Симптомите зависят от това кои части на мозъка са значително увредени. Състоянието може да се класифицира като семантично, нефлуентно аграматично или логопедично в зависимост от преобладаващите дефицити на пациента.
Първично прогресивната афазия няма лечение и състоянието прогресира с времето. Въпреки това, в зависимост от подтипа на заболяването и индивидуалните фактори, подходящият подход при терапия може да помогне за поддържане на функционалните способности и да подобри качеството на живот на пациентите и лицата, които се грижат за тях.
История
Общата концепция за първично прогресиращ болестен процес на езиково увреждане без когнитивни ефекти е описана за първи път през 1890 година от Pick и Serieux.
Заболяването е описано първи път като отделен синдром от M. Marsel Mesulam през 1980 година и дава името "бавно прогресиращата афазия". По-късно е преименувано в литературата на „първично прогресивна афазия“.
Епидемиология
Разпространението на заболяването се оценява на 3 до 4 на 100 000 население. Честотата на това състояние е около 1,14 на 100 000 човека, в сравнение с 35,7 на 100 000 човека за болестта на Алцхаймер.
Разпространението на нефлуентния вариант е приблизително 0,5 до 3,9 на 100 000 души. Мъжете и жените са еднакво засегнати, със средна възраст около 60 години.
Не са идентифицирани демографски, екологични или социално-икономически рискови фактори.
Приблизително 56% до 86% от хората имат съпътстваща говорна апраксия. Скорошно проучване показа, че поведенческият вариант е по-чест при мъжете, докато първичната прогресивна афазия е по-честа при жените.
Етиология
Първичната прогресивна афазия е група от невродегенеративни заболявания, засягащи предимно езиковите (доминиращи) области на мозъка. Разстройството обикновено се счита за подтип на фронтотемпоралната деменция (ФТД). От 2011 година ФТД се класифицира клинично и патологично в по-често срещания поведенчески вариант и първично прогресивна афазия.
Трите варианта на ППА възникват от невродегенеративни промени в различни мозъчни области. Семантичните и нефлуентните варианти са свързани с ФТД, докато логопедичният тип прилича повече на болестта на Алцхаймер. Семантичният вариант засяга главно предната времева област. Нефлуентният вариант засяга областите на прецентралната кора. Логопедичният вариант включва темпоралната област.
Нефлуентен или аграматичен вариант
Най-често се свързва с FTD-4R тау протеинова форма. Други доклади показват патология на TDP-43-A.
Някои случаи са свързани с генни мутации на програнулин или промени в хромозома 9.
Често се наблюдава фронтотемпоралната лобарна дегенерация.
Семантичен вариант
Почти винаги се свързва с подлежащи TDP-43-C патологични агрегати, открити в 75–100% от случаите в клинико-патологичните корелационни серии. Тази форма се свързва най-често с патология на левия (доминиращ) темпорален лоб.
Логопедичен вариант
Най-често се причинява от патология, свързана с болестта на Алцхаймер в цели 95% от случаите. Това състояние се признава за едно от потенциалните фокални и ранни прояви на болестта на Алцхаймер, въпреки че други патологични профили като деменция с телца на Lewy, TDP-43 и тау се съобщават по-рядко.
Рискови фактори
Не са известни рискови фактори от околната среда за прогресивните афазии. Проучване предполага, че вазектомията може да бъде рисков фактор за ППА при мъжете.
Първичната прогресивна афазия не се счита за наследствено заболяване. Въпреки това, роднините на човек, с каквато и да е форма на фронтотемпорална лобарна дегенерация, включително ППА, са изложени на малко по-голям риск от развитие на първична прогресивна афазия или друга форма на заболяването.
Патофизиология
Патологията, лежаща в основата на семантичната форма, включва главно атрофия и хипометаболизъм на предните темпорални лобове, с изразено левостранно преобладаване. В 80% от случаите се установяват агрегати от TAR ДНК-свързващ протеин 43kDa (TDP-43), подобно на патологичните характеристики, наблюдавани при фронтотемпоралната деменция и амиотрофична латерална склероза.
При нефлуентната форма се наблюдава засягане на доминиращия долен фронтален лоб (зоната на Broca), като атрофията на кортекса в доминантното полукълбо също е често срещана. Променливостта на това състояние съвпада с регионите на мозъка, свързани с речта и формирането на изреченията. Много пациенти проявяват абнормено отлагане на тау, докато при по-малка част може да се установи TDP-43 или патология на Алцхаймер.
Логопедичната форма най-често е афазичен вариант на болестта на Алцхаймер. Пациентите най-често развиват типичните клинични характеристики на болестта на Алцхаймер, но с повече левостранна атрофия. Подобно на болестта на Алцхаймер също има натрупване на амилоид и хиперфосфорилиран тау.
Класификация
Първичната прогресивна афазия тясно свързано с болестите на Alzheimer и Pick. По този начин класификацията и диагнозата на PPA понякога могат да бъдат противоречиви. Трите основни варианта на PPA са:
- нефлуентен/аграматичен (nfvPPA)
- семантичен (svPPA)
- логопедичен (lvPPA)
Първичната прогресивна афазия се проявява подобно на първичната прогресивна апраксия - постепенно развиващо се дегенеративно разстройство, засягащо планирането и изпълнението на сензомоторни команди, необходими за формирането на речта. Патологично, nfvPPA е групиран със синдромите на фронтотемпорална деменция. За разлика от това, логопедичният вариант често се класифицира като атипична форма на болестта на Алцхаймер.
За първия вариант основните критерии за диагноза включват аграматизъм и бавна и затруднена реч. Непоследователните звукови грешки в говора също са много чести, включително изкривявания, пропускане на звуци и вмъквания. По отношение на разбирането има дефицити в синтаксиса и разбирането на изреченията поради граматическа сложност, но разбирането на една дума е възможно.
Вторият вариант се представя с дефицити в разбирането на едносрична дума и предмет. Уврежданията при именуване могат да бъдат тежки и в крайна сметка могат да доведат до по-разпространен дефицит на семантична памет с течение на времето. Способността за четене и писане също може да бъде нарушена, ако има нередности между произношението и правописа. Въпреки това, повторението и моторната реч са относително запазени.
Логопедичният вариант включва нарушения в произнасянето на думи, повторение на изреченията и фонологични парафазии, сравними с кондуктивната афазия. В сравнение със семантичния вариант, разбирането и назоваването на една дума е запазено, но разбирането на изречението представлява трудност поради дължината и граматическата сложност. Речта включва незавършени думи, колебания, предшестващи думите, и повторения.
Симптоми и признаци при Ограничена атрофия на главния мозък МКБ G31.0
- Усещане за отпадналост
- Гърчове
- Загуба на тегло
- Намален мускулен тонус
- Симптоми на речта
- Поведенчески симптоми
Коментари към Ограничена атрофия на главния мозък МКБ G31.0