Други комбинирани имунодефицити МКБ D81.8
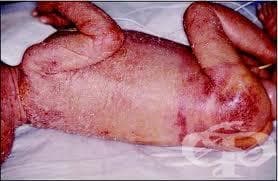
Синдромът на Omenn е форма на тежък комбиниран имунодефицит, която се характеризира с повишена предразположеност към опортюнистични инфекции, еозинофилия, еритродермия, алопеция, хронична диария, изоставане в растежа, лимфаденопатия и хепатоспленомегалия. Известен е още като комбиниран имунодефицит с хипереозинофилия.
Заболяването се унаследява по автозомно-рецесивен път. Болните бебета страдат от чести гъбични, бактериални или вирусни инфекции, които са типична проява на всеки комбиниран имунодефицит.
Открити са мутации на гените RAG-1 и RAG-2. Синдромът на Omenn понякога се дължи на мутации на други гени, освен това са описани случаи на делеция на 22 хромозома.
Имунологично се установява характерна липса на В-клетки при наличие на Т-лимфоцити. Липсата на В-лимфоцити обуславя тежка хипогамаглобулинемия с ниски нива на имуноглобулините от клас IgG. Наблюдаваната лимфоцитоза е резултат от пролиферацията на антиген - стимулирани Т-хелперни клетки, които произвеждат в голямо количество IL-4, IL-5. Тези цитокини провокират развитието на еозинофилия и повишен IgE.
Поради автозомно-рецесивното унаследяване, синдромът на Omenn се среща еднакво при представителите на двата пола.
Клинично, седмици след раждането, заболяването се демонстрира с еритродермия и диария. Бебетата често имат сепсис, дължащ се на S. aureus, който е свързан с генерализирания дерматит. Той често се бърка с екзема и прогресира до десквамация на кожата - отпадане на повърхностните слоеве. По този начин организмът губи протеини и възникват отоци.
Хроничната диария води до изоставане в растежа на децата. Макар да не е характерна проява за другите форми на комбиниран имунодефицит, при синдрома на Omenn се развива генерализирана лимфаденопатия и хепатоспленомегалия. Други прояви са пневмонии, предизвикани от Pneumocystis carinii, полиомиелит, както и алопеция.
Единственото дефинитивно лечението е трансплантацията на костен мозък. Ако не се извърши, бебетата умират в първите няколко месеца от живота си.
Друга терапия - имуносупресивни лекарства, за да се контролира Т-клетъчната пролиферация, антибиотици, локални кортикостероидни кремове, адекватно хранене - за компенсиране на диарията.
Дефицит на биотин-зависима карбоксилаза се характеризира с дефицит на четири известни биотин-зависими ензими, описани при човека. Тези ензими са ацетил КоА карбоксилаза - основен при синтезата на мастни киселини, пироват карбоксилаза - катализира началния етап в глюконеогенезата, пропионил КоА карбоксилаза - катаболизира разклонено-верижните аминокиселини валин, изолевцин, метионин и треонин, и бета-метил кротонил КоА карбоксилаза, която катализира катаболизма на левцин.
Биотинът е коензим на карбоксилазите, участващи в синтеза на мастни киселини, изолевцин, валин, в глюконеогенезата и е необходим за растежа на клетките. Той играе важна роля в цикъла на лимонената киселина, която е биохимичен процес, чрез който се генерира енергия по аеробен път. Биотин не само участва в различни метаболитни реакции, но също така помага за прехвърлянето на въглероден диоксид и поддържането на стабилно ниво на кръвната захар.
Други комбинирани имунодефицити включват дефицит на биотин-зависима карбоксилаза. Описани са изолирани дефицити на всяка от четирите биотин-зависими карбоксилази. Множественият карбоксилазен дефицит обединява наследствени метаболитни заболявания, характеризиращи се с дефицит на биотин-зависими карбоксилази.
Биохимичните и клинични прояви включват: лактатна ацидоза, органична ацидурия, хиперамонемия, кожен обрив, проблеми с храненето, хипотония, гърчове, изоставане в развитието, алопеция и кома. Симптомите се появяват в ранна детска. Чести прояви с тежко протичащи инфекции, кандидозен дерматит, кератоконюнктивит и интермитентна атаксия. При част от пациентите, в резултат на усложнения от страна на централната нервна система и тежки инфекции, се стига до фатален изход.
Имунологичните нарушения се характеризират с липса на отговор при кожен тест за забавена свръхчувствителност и ин витро, установена липса на лимфоцитен отговор към антигени на кандида. Установен е селективен IgA дефицит, липса на антителен отговор при имунизация срещу пневмококи и намален брой на Т-лимфоцитите в периферната кръв.
Лабораторните изследвания установяват лактатна ацидоза и повишена екскреция на β-хидроксипропионат, метилцитрат, β-метилкротонилглицин, β-хидроксиизовалерат с урината.
Лечение с перорален биотин 10 мг/дневно, води до значително понижение на метаболити в урината, което предполага, множествен карбоксилазен дефицит, отговарящ на терапия с биотин.
Симптоми и признаци при Други комбинирани имунодефицити МКБ D81.8
- Увеличен черен дроб
- Намален мускулен тонус
- Увеличен далак
- Липса на растеж
- Симптоми на дихателната система
- Инфекция
Коментари към Други комбинирани имунодефицити МКБ D81.8