Други ганглиозидози МКБ E75.1
Към други ганглиозидози принадлежат ганглиозидоза GM1, ганглиозидоза GM3 и муколипидоза 4.
ганглиозидоза GM1
Ганглиозидозите GM1 (монозиалотетрахексозилганглиозид), ганглиозидози, причинени от мутация в гена GLB1, водеща до дефицит на бета-галактозидаза.
Дефицитът причинява нарушения в съхранението на киселинни липидни вещества в клетките на централната и периферната нервна система, но особено в нервните клетки, което води до прогресивна невродегенерация.
GM1 ганглиозидозата е рядко лизозомно нарушение с разпространение от 1 на всеки 100 000 до 200 000 живородени по света, въпреки че процентите са по-високи в някои региони.
Етиология
GM1 ганглиозидозните нарушения се причиняват от мутации в гена GLB1, който кодира лизозомна хидролаза, кисела бета-галактозидаза (β-gal). Ниските нива на β-gal причиняват натрупване на GM1 ганглиозиди.
Това са наследствени, автозомно-рецесивни сфинголипидози, свързани с нарушения в натрупването на липиди.
Класификация
GM1 има три форми, класифицирани според възрастта на поява.
- Тип 1: ранна инфантилна
- Тип 2: късна инфантилна/ювенилна
- Тип 3: възрастна
Клинична картина
Ранна инфантилна форма
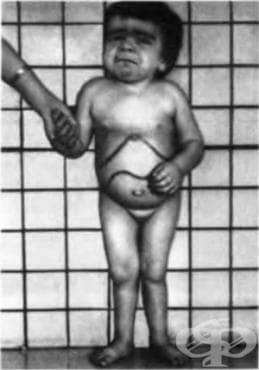
Симптомите на тази форма (най-тежкият подтип, с начало малко след раждането) могат да включват невродегенерация, гърчове, уголемяване на черния дроб (хепатомегалия), уголемяване на далака (спленомегалия), загрубяване на чертите на лицето, скованост на ставите, подуване на корема, мускулна слабост, изразена проява на стрес при звук и проблеми с походката.
Децата могат да бъдат с нарушени слух и зрение до 1-годишна възраст и често умират до 3-годишна възраст от сърдечни усложнения или пневмония.
Децата могат да имат увреждания на слуха и зрението преди навършване на 1 година и често умират преди 3-годишна възраст от сърдечни усложнения или пневмония.
Ранни психомоторни нарушения:
- намалена активност и летаргия през първите седмици;
- проблеми с храненето - нарушен растеж;
- зрителни нарушения (нистагъм) до 6 месеца;
- хипотония;
- спастичност с пирамидални признаци;
- развитие на вторична микроцефалия;
- децеребрална ригидност до 1 година и смърт до възраст 1-2 години (поради пневмония и дихателна недостатъчност);
Установяват се макулни вишнево-червени петна в 50% от случаите до 6-10 месеца.
Тази форма се характеризира и с лицев дисморфизъм:
- изпъкнало чело
- широк нос
- оток на лицето (подпухнали клепачи)
- периферен оток
- дълга горна устна
- микроретрогнатия
- гингивална хипертрофия (дебели алвеоларни ръбове)
- макроглосия
Описана е хепатомегалия до 6 месеца и спленомегалия по-късно. В някои случаи е налице сърдечна недостатъчност.
Ранната форма се характеризира още и със скелетни деформации:
- флексионни контрактури, забелязани след 3 месеца
- ранно образуване на субпериостална кост (може да присъства при раждането)
- разширяване на диафизата
- деминерализация
- тораколумбална вертебрална хипоплазия на възраст 3–6 месеца
- кифосколиоза.
Между 10 и 80% от периферните лимфоцити са вакуолизирани, налице са и пенести хистиоцити в костния мозък;
Нивото на GM1 в мозъчното сиво вещество е 10-кратно повишено.
Налице е галактоза-съдържаща олигозахаридурия и умерена кератансулфатурия
Късна инфантилна/ювенилна форма
Началото на този вариант обикновено е на възраст между 1 и 3 години. Ювенилната форма може да бъде диагностицирана в детска възраст.
Някои деца доживяват до юношеска възраст или ранна зряла възраст. Този подтип се характеризира с придобиване на някои признаци на развитие, след което настъпват забавяния, последвани от регресия.
Ранните симптоми включват затруднено пълзене и ходене, хипотония, проблеми с говора и преглъщането, както и гърчове. Неврологичните симптоми включват атаксия, гърчове, деменция и затруднения с говора.
Възрастна форма
Началото на GM1 при възрастни обикновено е в юношеска или зряла възраст и е най-бавно прогресиращият от подтиповете.
Симптомите включват мускулна атрофия, неврологични усложнения, които са по-леки и прогресират с по-бавна скорост, отколкото при други форми на заболяването, помътняване на роговицата при някои пациенти и дистония (продължителни мускулни контракции, които причиняват усукване и повтарящи се движения или необичайни пози).
В долната част на тялото на тялото могат да се развият ангиокератоми.
Повечето пациенти имат нормален размер на черния дроб и далака. Пренаталната диагностика е възможна чрез измерване на бета галактозидаза в култивирани амниотични клетки.
Диагноза
Диагнозата на GM1 може да се постави чрез генетично и ензимно изследване.
Лечение
Лечението на GM1 е базирано на симптомите и е палиативно. Няма лек за GM1, въпреки че са в ход няколко проучвания за генна терапия.
Към други ганглиозидози принадлежи ганглиозидоза GM3.
Ганглиозидоза GM3 представлява заболяване, което се дължи на разстройство на ганглиозидната биосинтеза, причинено от (N-acetylneuraminyl)-galactosylglucosylceramide N-ацетил трансфераза вследствие на което се получава прекомерно натрупване на ганглиозид GM3 в черния дроб и мозъчна тъкан в отсъствието на по-високи хомолози, ганглиозиди.
Клиничните характеристики на заболяването са много специфични и включват изтощеност, забавено психомоторно развитие, загрубяване на чертите на лицето, макроглосия, хипертрофия на венците, хепатоспленомегалия, ингвинална херния наличие на къси и дебели ръце и крака. Повечето пациенти умират в ранна детска възраст.
Диагнозата на ганглиозидоза GM3, се потвърждава при провеждането на специфични изследвания, клиничната картина, прегледа от лекар специалист.
Към други ганглиозидози принадлежи муколипидоза тип 4.
Муколипидоза тип IV (MLIV) е рядко автозомно-рецесивно заболяване, причинено от мутации в MCOLN1. Този ген кодира муколипин-1 - протеин с неизвестна функция, принадлежащ към семейството на гените на преходния рецепторен потенциал (TRP), обикновено означаван като TRPML1.
Проучванията показват, че протеинът има функция на катионен канал в клетките и безклетъчните системи, което е подобно на функцията на TRP каналите.
Промените в муколипина водят до натрупване на хетерогенни липиди и протеини в цитоплазмени вакуоли, получени от лизозоми. Обратно, други муколипидози с липидни включвания се причиняват от анормални метаболитни ензими и включват муколипидоза I (сиалидоза), причинена от мутирала сиалидаза и муколипидоза II (I-клетъчно заболяване) и тип III (псевдо-Hurler полидистрофия), и двете в резултат на мутации в гена за N-ацетилглюкозамин-1-фосфотрансфераза.
Обикновено заболяването започва в ранна детска възраст, като се проявява с тежко психомоторно забавяне, зрително увреждане и ахлоридрия. След началото на заболяването често настъпва период на стабилизиране, който продължава две до три десетилетия.
История
Муколипидоза 4 (MLIV) е описана за първи път през 1974 година при ешкеназко еврейско бебе, което се проявява с двустранно помътняване на роговицата, леко забавяне на развитието и цитоплазмени включвания в култивирани клетки от черния дроб, конюнктивата и кожните тъкани.
Епидемиология
Муколипидоза 4 е пан-етническо разстройство заболяване, идентифицирано по целия свят, въпреки че приблизително 70-80% от описаните пациенти имат еврейско потекло от Ашкенази.
По-голямата част от първите 100 описани пациенти с MLIV са евреи ашкенази с полско или литовско потекло. Идентифицирането на два различни хаплотипа води до последващ анализ на връзката и в крайна сметка допринася за откриването на гена, причиняващ заболяване.
Изчислено е, че 1 на 100 ашкеназки евреи са носители на един от тези два хаплотипа, с честота на заболяването 1 на 40 000.
Три други лизозомни болести на натрупването често се срещат в еврейското население на Ашкенази: болест на Gaucher, болест на Tay-Sachs и тип А и В на Niemann-Pick.
Като цяло лизозомните нарушения се срещат при приблизително едно на всеки 7700 живородени.
Патоморфология
Клетките на пациенти с MLIV съдържат разтворими протеинови и липидни агрегати. Фоспохолипиди, мукополизахариди и ганглиозиди се намират в тези включени тела. Тези телца съдържат хетерогенна гама от липиди във всички клетки, с изключение на ЦНС, където тези включвания се характеризират с големи количества липиди, получени от сиалова киселина.
Аномалиите са подобни на тези, наблюдавани в клетки на пациенти с GM2-ганглиозидоза тип I или болест на Tay-Sachs.
Клинична картина
Системни прояви
По-голямата част от пациентите с муколипидоза 4, докладвани в литературата, се считат за „типичен MLIV“. Тежкото когнитивно увреждане и очните аномалии са отличителни симптоми на типичната форма. Ахлорхидрия, която води до повишени нива на гастрин в кръвта, и желязодефицитна анемия също са чести прояви.
Сред най-често срещаните симптоми на MLIV са психомоторни забавяния, помътняване на роговицата, дистрофия на ретината, оптична атрофия и страбизъм.
Пациентите с това заболяване са в долните персентили за височина, тегло и обиколка на главата. Бременностите и перинаталният период на бебета с MLIV обикновено са нормални, със средно тегло при раждане, въпреки че впоследствие физическият растеж е силно забавен с течение на времето. Проявите при раждането на някои пациенти обаче предполага пренатално начало на заболяването.
Дисморфичните характеристики, въпреки че често се наблюдават при пациенти с други муколипидози, не са характерни за пациенти с MLIV.
Неочаквано пациентите с MLIV имат нормални нива на мукополизахариди, олигозахариди и лизозомни хидролази в кръвта. Това е разликата от повечето лизозомни болести на натрупването, които се дължат на аномалии в лизозомните хидролази или свързаните с тях лизозомни разграждащи ензими.
Въпреки че лизозомни включвания се откриват в повечето тъкани, включително черен дроб, бъбрек, кожа и далак, органомегалията не е характеристика на MLIV. Съдържанието на липиди и протеини в тези цитозолни включвания е хетерогенно и съставът може да варира между видовете тъкан, вероятно поради разликите в лизозомната дисфункция между видовете тъкани.
Очни прояви
При повечето пациенти двустранното помътняване на роговицата и страбизмът са първите забележими признаци на заболяването. Те могат да бъдат забелязани още при раждането и обикновено се наблюдават през първата и втората година от живота.
Непрозрачността на роговицата се счита за отличителна находка. Въпреки това, помътняване на роговицата може да настъпи и на 13-годишна възраст при някои пациенти. Освен това, в някои случаи е отбелязано двигателно забавяне преди началото на помътняването на роговицата.
Въпреки наблюдаваната стабилност на заболяването през второто и третото десетилетие от живота, офталмологичното засягане обикновено прогресира.
Тежестта на офталмологичните аномалии при по-големи деца може да бъде повлияна от хормонални промени в пубертета или от кумулативните ефекти на клетъчни лизозомни включвания.
Често дегенерацията на ретината, а не помътняването на роговицата, е причина за прогресирането на зрителното увреждане. Смята се, че такава дегенерация се дължи на включвания в клетките на ретината.
Значително намалено зрение възниква, когато съдовото отслабване на ретината, промените в пигментацията на ретината и дисплазията на роговицата са тежки. Освен това някои пациенти изпитват силна очна болка, сълзене и ипсилатерално зачервяване на лицето, което може да доведе до фотофобия. В крайна сметка може да настъпи слепота.
Електроретинографията (ERG) показва отклонения при пациенти с MLIV.
Неврологични прояви
Съобщава се за невродегенерация при повечето пациенти с това заболяване. Спастичността, хипотонията и неспособността за самостоятелно ходене са чести сред такива пациенти и обикновено започват в ранна детска възраст.
Неврологичните симптоми понякога се разпознават при кърмачета, представяйки се като забавено постигане на основните етапи на общата моторика. В повечето случаи развитието на езиковите и двигателните функции никога не надхвърля 12-15 месеца.
При някои пациенти се съобщава за бавно, но непрекъснато подобрение на когнитивните, езиковите и двигателните функции.
Птоза, миопатичен фациес, постепенно намаляване на движенията на лицето, постоянно слюноотделяне и затруднения при дъвчене и преглъщане също са докладвани и се смята, че се дължат на засягане на черепните нерви.
Електроенцефалографските (ЕЕГ) записи показват, че епилептични разряди често се откриват при пациенти с MLIV, но те рядко са свързани с клинични припадъци.
Изследванията с ядрено-магнитен резонанс (ЯМР) разкриват промени в corpus callosum, дисмиелинизация на бялото вещество, намален интензитет на сигнала в базалните ганглии и церебеларна атрофия. Церебеларната атрофия обикновено се среща при по-възрастни пациенти.
Диагноза
Измерването на нивата на гастрин в кръвта е полезен метод за диагноза. Пациентите с MLIV средно имат нива на гастрин в диапазона от 400-4100 pg/ml (средно 1507 pg/ml) в сравнение с диапазон от 0-200 pg/ml при нормални контроли.
Освен това, ядрено-магнитен резонанс (ЯМР), показващ тънък corpus callosum, може да покаже промени, насочващи към заболяването.
Кожните биопсии често се използват за потвърждаване на диагнозата]. Култивираните фибробласти показват мембранно- и не-мембранно-свързани цитоплазмени включвания, които могат да се наблюдават под електронен светлинен микроскоп. Мембраните изглеждат ламинирани, докато разтворимият материал е аморфен и гранулиран.
Като алтернатива може да се използва киселинно-шифово оцветяване на епителните клетки, за да се покажат типичните аморфни включвания, които също са автофлуоресцентни.
Пренаталната диагностика е възможна с помощта на електронна микроскопия, въпреки че диагнозата обикновено се потвърждава чрез скрининг за мутации. Въпреки това, скринингът за най-честите мутации при пациенти от неашкеназки евреи може да демонстрира фалшиви отрицателни резултати и по този начин по-изчерпателният генетичен скрининг е жизненоважен за установяване на диагнозата.
Лечение
Няма специфично лечение за това състояние.
Терапията за някои от лизозомните разстройства включват ензимна заместителна терапия, субстрат редукционна терапия и генна терапия.
Физикалната терапия със специален фокус върху спастичността и атаксията може да подобри двигателната функция. Някои от двигателните нарушения могат да бъдат коригирани с ортопедична намеса.
Добавките с желязо се използват за лечение на желязодефицитна анемия, която често е резултат от лошо усвояване на хранителни вещества от стомашната лигавица.
Продължително носене на терапевтични меки контактни лещи може да подобри комфорта на очите. Трансплантацията на роговицата и конюнктивата се използва за коригиране на зрителното увреждане, въпреки че присадената тъкан развива аномалии, подобни на тези на тъканта на гостоприемника.
Коментари към Други ганглиозидози МКБ E75.1