Други мукополизахаридози МКБ E76.2
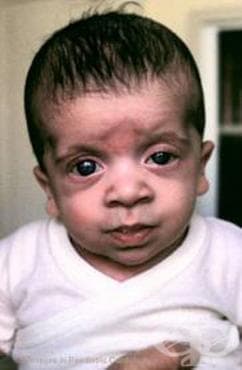
Други мукополизахаридози представлява група от рядко срещани заболявания.
Към други мукополизахаридози принадлежат недоимъкът на β-глюкуронидаза, мукополизахаридоза тип 3,4,6,7, синдром на Maroteaux-Lamy, Marquio, Sanfilipo.
Към други мукополизахаридози принадлежи недоимъкът на β-глюкуронидаза.
Бета-глюкоронидазите са членове на семейството на гликозидазните ензими, които катализират разграждането на сложни въглехидрати. Човешката β-глюкуронидаза е вид глюкуронидаза, която катализира хидролизата на β-D-глюкуронова киселина. Човешката β-глюкуронидаза се намира в лизозомите в червата. Бета-глюкуронидазата също присъства в майчиното мляко, което допринася за неонатална жълтеница.
Недоимъкът на β-глюкуронидаза представлява не рецесивно наследствено метаболитно заболяване, известно като синдром на Слай или Мукополизахаридоза VII.
Недостигът на този ензим води до натрупването на не-хидролизирани мукополизахариди в пациента. Това заболяване може да бъде изключително инвалидизиращо за пациента или може да доведе до хидропс феталис преди раждането. В допълнение, умствена изостаналост, нисък ръст, груби черти на лицето, гръбначни аномалии и уголемяване на черния дроб и слезката.
За диагнозата основно значение имат клиничната картина, анамнезата, специализираните специфични изследвания и лабораторните изследвания.
Диференциална диагноза се прави с всички заболявания, които имат подобна клинична симптоматика.
Лечението на заболяването е специфично и се осъществява от лекар специалист.
Към други мукополизахаридози принадлежи синдрома на Санфелипо (Sanfilipo).
Санфелипо (Sanfilippo) синдром или мукополизахаридоза тип III представлява рядко автозомно-рецесивно лизозомно заболяване.
Заболяването се причинява от дефицит на един от ензимите, необходими за разграждането на гликозаминогликанова хепаран сулфат (който се намира в извънклетъчната матрица и гликопротеини на клетъчната повърхност).
Заболяването е описано от Силвестър Санфелипо (Sanfilippo), педиатър който пръв описва това заболяване.
Честотата на синдрома Санфелипо (Sanfilippo) варира географски, с приблизително 1 случай на 280 000 живородени деца в Северна Ирландия, 1 на 66 000 в Австралия и 1 на 50 000 в Нидерландия.
Заболяването се проявява при малки деца.
Засегнатите деца са очевидно нормални, въпреки че при някои се наблюдава лек лицев дисморфизъм. Скованост на ставите, наличие на груби косми, характерни за други мукополизахаридози обикновено не се наблюдават до напредванетов на болестта.
При болните обикновено се наблюдава забавяне на развитието и / или поведенчески проблеми, последвано от постепенен интелектуален спад в резултат на тежката деменция.
Заболяването прогресира до увеличаване на поведенчески смущения, включително изблици на раздразнение, хиперактивност, разрушителност, агресивност и нарушения на съня.
В последната фаза на заболяването деца стават все по-неподвижни, често изискват инвалидни колички. Продължителност на живота на засегнатото дете обикновено не се простира отвъд края на пубертета до началото на двадесетте години.
Въпреки че клиничните характеристики на заболяването са предимно неврологични, при част от пациентите могат да се наблюдават симптоми като диария, кариозни зъби, както и увеличение на черния дроб и далака.
От всички мукополизахаридози, мукополизахаридоза тип III се характеризира с най-леките физически аномалии.
Диагнозата на заболяването може да се потвърди чрез анализ на ензимните нива в тъканни проби и генно секвениране. Възможна е и пренатална диагностика.
Лечението е трудно и се осъществява от лекар специалист.
Към други мукополизахаридози принадлежи синдрома на Марото Лами (Maroteaux-Lamy).
Марото Лами синдром (известен също като мукополизахаридоза тип VI или полидистропичен нанизъм) е форма на мукополизахаридоза, която се дължи на дефицит на арилсулфатаза. Заболяването е описано за пръв път от Пиер Марото през 1926 г.
Децата със синдром на Марото Лами обикновено имат нормално интелектуално развитие, но имат много от физическите симптоми, които се наблюдават при синдрома Hurler. Неврологични усложнения се изразяват в потъмняване на роговица, глухота, удебеляване на обвивката (мембрана, която заобикаля и защитава мозъка и гръбначния мозък) и болка, която се дължи на засягане на нервните окончания.
Признаците характерни за заболяването се наблюдават още от началото на живота на засегнатото дете, като един от първите симптоми, който най-често се наблюдава е значително удължена възраст на прохождане. При по-тежките случаи, децата имат тежки скелетни аномалии. При не малка част от деца се наблюдава пъпна или ингвинална херния. Почти всички деца имат някаква кардиопатия, като най-често става на въпрос за клапна дисфункция.
Ензим-заместваща терапия при пациенти с Марото-Lamy синдром и бе успешна и се наблюдава значително подобряване състоянието на пациентите.
Диагнозата на заболяването може да се потвърди чрез анализ на ензимните нива в тъканни проби и генно секвениране. Възможна е и пренатална диагностика.
Лечението е трудно и се осъществява от лекар специалист.
Към други мукополизахаридози принадлежи синдрома на Моркио (Marquio).
Синдром на Моркио (известен още като мукополизахаридоза тип IV) е автозомно-рецесивно заболяване. Това е рядък вид вроден дефект със сериозни последици. Когато тялото не може да обработва някои видове мукополизахариди, те повишават или се елиминират по начин, който води до различни симптоми
Пациентите със синдром на Моркио изглеждат здрави при раждането.
Със синдрома на Моркио са свързани следните симптоми нарушено сърдечно развитие, ненормално развитие на скелета, повишена ставна мобилност, наличие на големи пръсти, обърнати навътре колене, широко разположени зъби, компресия на гръбначния мозък, уголемено сърце, сърдечни шумове, нисък ръст.
Диагнозата на заболяването може да се потвърди чрез анализ на ензимните нива в тъканни проби и генно секвениране. Възможна е и пренатална диагностика.
Лечението на синдрома Morquio се състои от пренатална идентификация и ензим-заместваща терапия.
Коментари към Други мукополизахаридози МКБ E76.2