Ендогенни пигменти
Ендогенните пигменти по своята химическа природа представляват хромопротеиди. Те изпълняват разнообразни функции. Така например с помощта на хемоглобина, миоглобина и цитохромите се осъществява свързването и пренасянето на кислорода до клетките и тъканите. Меланинът защитава кожата от въздействието на ултравиолетовите лъчи.
Натрупването на ендогенни пигменти може да бъде в резултат на генетично обусловени причини или вторично провокирано от някое заболяване. В зависимост от своя произход се различават три типа ендогенни пигменти:
- хемоглобиногенни - феритин, хемосидерин, билирубин
- тирозиногенни - меланин
- липидогенни
При нормални условия се образуват следните пигменти поради физиологичното разграждане на еритроцитите и хемоглобина: феритин, хемосидерин, билирубин. При патологични състояния, когато разграждането на еритроцитите се увеличава, се синтезират нови пигменти, с изключение на увеличаването на количеството на горните пигменти. Те са хематодин, хематини, порфирин.
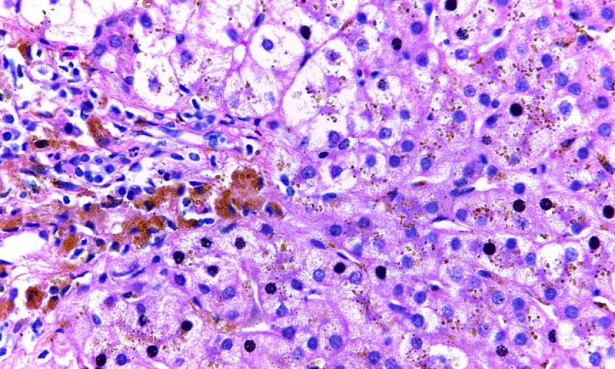
Най-важното нарушение е хемосидерозата, тоест изобилната продукция на хемосидерин.
Желязото може да се натрупва в черния дроб при много различни условия, от които клинично най-важната е наследствената хемохроматоза. За анатомичния патолог разграничението между натрупването на желязо и други причини може да бъде сложно и успехът често зависи от комбинация от морфологични наблюдения, клинична информация, генетични изследвания и / или количествен анализ на желязото в черния дроб. Самото наличие на гранулирано желязо в черния дроб се нарича "хемосидероза" - това представлява морфологично наблюдение, а не специфично заболяване.
Хемохроматозата е болестно състояние, характеризиращо се с отлагането на хемосидерин, но е станало ясно, че съществуват както възрастни, така и млади форми на хемохроматоза и че са замесени герминативни мутации на най-малко четири различни гена.
През 1889 година фон Реклингхаузен идентифицира излишното количество желязо от тъкани, получено при аутопсия, и наречено състоянието "хемохроматоза". Това желязо се смята за произхождащо от кръвообращението до 1935 година, когато британския геронтолог Шелдон публикува своите открития от 311 избрани пациенти, заключавайки, че натрупването на желязото е вторично, поради увеличената абсорбция на желязо.
Тип 1 хемохроматоза е най-добре изследваната форма и е типът, свързан с мутациите в гена на HFE. Ясно е, че основната аномалия, присъстваща в това състояние, е прекомерната абсорбция на желязо в дванадесетопръстника. Независимо от точния механизъм при хемохроматоза тип 1 за възрастни, резултатът е прогресивно увеличаване на общото желязо в организма, което се отразява от увеличаването на наситеността на серумния трансферин и феритин и води до ненормално отлагане на желязо, най-вече в хепатоцитите.
Тежко усложнение, което се среща рядко, е прогресивната чернодробна фиброза и в крайна сметка цироза, придружени от повишен риск от хепатоцелуларен карцином. По-честите симптоми включват дифузна артропатия и генерализирана умора. Сърдечните заболявания и сърдечната недостатъчност се наблюдават при малцинство пациенти, но са доминиращи фактори в редките млади форми на хемохроматоза. Също така се наблюдават различни ендокринни разстройства - захарен диабет, хипогонадизъм и хипотиреоидизъм. Рядко се наблюдава кожна пигментация ("бронзов диабет"). По-слабо характеризираните са потенциалните повишени рискове от екстрахепатален злокачествен инцидент и за инфекции със системно претоварване с желязо.
Младите форми на наследствена хемохроматоза са много по-рядко срещани в сравнение с възрастовите форми. Неонаталната хемохроматоза не е наследена и остава с несигурна патогенеза. Може да се появи в края на втория или третия триместър като загуба на плода или в часовете до седмици след раждането като остра чернодробна недостатъчност.
Отличителен белег на тип 1 хемохроматоза е отлагането на хемосидерин в хепатоцити и жлъчния епител. Хемосидеринът е неразтворим и се появи на гранули с железни петна. Алтернативно, желязо може да бъде депозиран под формата на феритин, който е разтворим и се характеризира с дифузно, светло синьо оцветяване на хепатоцитите и е много неспецифично.
В ранните фази на заболяването, хемосидеринът се депозира в перипорталните хепатоцити. Тъй като желязото се натрупва, евентуално отделни хепатоцити ще натрупват смъртоносни нива на желязо и ще претърпят "сидеронекроза". След това местно освободеното желязо се поема от макрофаги, но хепатоцелуларното желязо ще продължи да доминира. Макрофагичното желязо е свързано с прогресивна фиброза. Когато е налице цироза, тя е с лека природа с фини влакнести тъканни участъци, обграждащи регенеративните възли.
Хипербилирубинемията се отнася до повишена серумна концентрация на билирубин (тоест > 1.2 mg / dl). При концентрации на серумния билирубин, отнасящ се към ендогенни пигменти, над 2 до 2,5 милиграма /децилитър, кожата, склериите, лигавиците и серумните мембрани стават жълти - състояние, известно като жълтеница. Според механизма на нейното развитие, има 3 вида жълтеница:
- Прехепатална (хемолитична, неконюгирана хипербилирубинемия) жълтеница - възниква в резултат на повишеното производство на билирубин при хемолиза на еритроцитите. Черният дроб произвежда повишено количество билирубин. Това нарушение се характеризира с повишени серумни нива на неконюгиран билирубин. Нивата на конюгирания билирубин са в нормални граници. Тази констатация характеризира състояния, свързани с увеличаване на разрушаването на червените кръвни клетки (например хемолитична анемия, неефективна еритропоеза), намалено поемане на чернодробен билирубин и нарушено конюгиране с билирубин. Това състояние се наблюдава при интоксикации, инфекции, автоимунни процеси. Това се дължи на невъзможност на хепатоцитите да конюгират билирубин и неспособността на билирубина да преминава от черния дроб в червата.
- Хепатоцелуларна (паренхиматозен) жълтеница се появява при хепатоцитно увреждане (остър и хроничен хепатит, цироза на черния дроб)
- Постхепатална (конюгирана хипербилирубинемия) или обструктивната жълтеница - резултат от обструкция на конюгиран билирубин (холелитиаза, рак на жлъчните пътища и други.). Нивата на конюгиран и неконюгиран билирубин се повишават в това заболяване. Вътрешното чернодробно заболяване и екстрахепаталната билиарна обструкция са основните причини за конюгираната хипербилирубинемия. Холестазата невинаги присъства.
Макроскопска картина: черният дроб нараства, придобива жълто-зелен цвят, вътрехепаталните канали се разширяват.
Микроскопски билирубинът се открива в жлъчните синусоиди, Купферовите клетки и хепатоцити под формата на зелени кафяви аморфни отлагания. Процесът започва в централните части на чернодробните лобули. При изразен процес се образуват големи агрегати на пигмента, водещи до фокална некроза на хепатоцитите. Екстрахепаталната обструкция се характеризира с оток на порталната строма с перидуктална левкоцитна инфилтрация, пролиферация на жлъчните пътища близо до граничната плака, постепенно развиваща се склероза.
Заглавно изображение: Calicut Medical College, CC BY-SA 4.0, via Wikimedia Commons
Библиография
http://www.nature.com/modpathol/journal/v20/n1s/fig_tab/3800715f4.html#figure-title
http://en.medicine-guidebook.com/patologicheskaya-fiziologiya_792_narusheniya-obmena-e~1.html
http://en.medicine-guidebook.com/patologicheskaya-fiziologiya_792_narusheniya-obmena-e~1.html
http://poznayka.org/s51658t1.html
Коментари към Ендогенни пигменти