Бързо прогресиращ гломерулонефрит
Бързо прогресиращият гломерулонефрит е клинично заболяване на бъбреците, характеризиращо се с бързо намаляване на скоростта на гломерулна филтрация от поне 50% за кратък период, от няколко дни до три месеца. Известен е още като полулунен гломерулонефрит и подостър гломерулонефрит. Основната патологична находка е образуването на гломерулен полумесец. Универсалната патологична особеност на бързо прогресиращ гломерулонефрит е фокално разкъсване на гломеруларните капилярни стени, които могат да се видят чрез светлинна микроскопия и електронна микроскопия.
Терминът бързо прогресиращ гломерулонефрит се използва за пръв път, за да опише група пациенти, които имат необичайно фулминантен постстрепококов гломерулонефрит и лош клиничен изход. В средата на 70-те години на миналия век е описана група пациенти, които отговарят на клиничните критерии за бързо прогресиращ гломерулонефрит, но при които не може да се установи причина. Много от тези случаи са свързани със системни признаци на съдово възпаление (системен васкулит), но някои случаи се характеризират само с бъбречно заболяване. Отличителна черта на тези случаи е виртуалната липса на отлагане на антитела след имунофлуоресцентно оцветяване на биопсичните проби. Над 80% от пациентите с бързо прогресиращ гломерулонефрит с имуносупресивен имунен отговор впоследствие са открили циркулиращи антинеутрофилни цитоплазмени антитела и следователно тази форма на бързо прогресиращ гломерулонефрит сега се нарича асоцииран с антинеутрофилни цитоплазмени антитела васкулит.
Лицата от бяла раса са засегнати по-често от чернокожите. Съотношението мъже/жени във всички проучвания е приблизително 1:1. Засегнатата възрастова група е 2-92 години. Въпреки това заболяването е рядкост при педиатричната популация. Върховата честота настъпва в средата на шестото десетилетие на живота.
Обикновено бързо прогресиращия гломерулонефрит възниква като усложнение на, след инфекциозен дифузен ендокапилярен гломерулонефрит или на системни заболявания (lupus erythematodes disseminatus, синдром на Goodpasture (Гудпасчър), васкулити (така наречения "микроскопски нодозен полиартериит", Вегенерова грануломатоза, пурпура на Shonlein - Henoch и есенциална криоглобулинемия), а в останалите случаи се разглежда като идиопатичен.
Бързо прогресиращият гломерулонефрит може да се класифицира в три типа, въз основа на моделите на имунофлуоресценция:
- Тип I - отчитането на приблизително 20% от случаите на бързо прогресиращ гломерулонефрит, тип I се характеризира с наличието на автоантитела, насочени срещу гломерулната основна мембрана (GBM). Той също така се нарича анти-GBM гломерулонефрит. Антителата са насочени срещу конкретен протеин, открит в гломерулната основна мембрана, тип IV колаген. В допълнение към анти-GBM антителата, някои случаи на тип I също са свързани с антитела, насочени срещу основната мембрана на белодробните алвеоли, произвеждащи синдром на Goodpasture. По-голямата част от заболяването тип I, обаче, се характеризира само с анти-GBM антитела. Тези случаи се считат за идиопатични.
- Тип II - се причинява от отлагането на имунни комплекси, представлява 25% от случаите на бързо прогресиращ гломерулонефрит. По този начин всяка имунна комплексна болест, която включва гломерула, може да се развие до бързо прогресиращ гломерулонефрит, ако е достатъчно тежка. Тези заболявания включват системен лупус еритематодес, остър пролиферативен гломерулонефрит, пурпура на Henoch-Schönlein.
- Тип III - представлява 55% от случаите на бързо прогресиращ гломерулонефрит и не притежава нито имуно-комплексно отлагане, нито анти-GBM антитела. Вместо това, гломерулите се увреждат по неопределен начин, може би чрез активиране на неутрофили в отговор на антинеутрофилни цитоплазмени антитела. Тип III може да бъде изолиран до гломерула (първичен или идиопатичен) или свързан със системно заболяване (вторичен).
В патофизиологията на бързо прогресиращия гломерулонефрит антинеутрофилните цитоплазмени антитела (ANCA) взаимодействат с антигени в цитоплазмата на неутрофилите. Смята се, че антинеутрофилните цитоплазмени антитела причиняват ранна дегранулация, даваща възможност за освобождаване на литични ензими в мястото на нараняване.
Макроскопски при бързо прогресиращ гломерулонефрит се установява, че двата бъбрека са силно увеличени и бледи - "големи бели бъбреци". Те са твърде отпуснати, за разлика от означаваната по същия начин находка при амилоидоза. На срез по-широката кора са пръснати жълти петна и ивички.
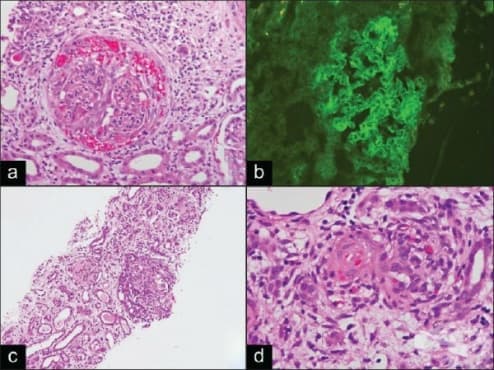
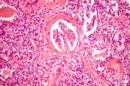
Микроскопски при бързо прогресиращ гломерулонефрит се установява, че в 70-80% от бъбречните телца се образуват екстра капилярни "полулуния" от струпвания на клетки в Баумановото пространство (Фигура 1). Те обхващат 2/3 от обиколката на Баумановата капсула и водят до склерозиране на гломерулите и бързо настъпваща бъбречна недостатъчност. При бързо прогресиращ гломерулонефрит полулунията са дифузни и с различна степен на зрелост. В гломерулите се установяват сегментни тромбози, огнищна фибриноидна некроза, левкоцитна инфилтрация и лека мезангиална хиперплазия. Капилярите са колабирани. Между тях и притискащите ги полулуния се намират множество синехии. В началото полулунията са "клетъчни", по-късно преминават във "фиброепителни". Процесът завършва с глобална склероза на бъбречните телца.
В някои случаи съдът може да покаже трансмурален васкулит, фибриноидна некроза, лимфоцитни инфилтрати и фрагментиран полиморф (левкоцитокластичен васкулит, фигура 2). Всеки съд може да участва, защото артериалните лезии са най-лесни за демонстрация. В интерстициалната област често се откриват възпалителни мононуклеарни инфилтрати, оток и напреднала фиброза. Тубулите могат да имат остри промени: тубулна некроза, епителна дегенерация и тубулит, или те могат да имат хронично увреждане - атрофия. При тип I, антителата могат също да разпознават антигени в тубуларни мембрани, които имат по-голямо тръбно увреждане и интерстициално възпаление. Интерстициалните грануломи, които не са свързани с гломерули, показват грануломатоза на Wegener, но това е много необичайна особеност в бъбреците. Те обикновено имат некротичен център и са заобиколени от епителиоидни хистиоцити и многоядрени гигантски клетки.
Белият дроб има обширен алвеоларен кръвоизлив, капилярно и вариращо интерстициално възпаление (както при тип I, така и при системен васкулит). Участието в други органи се характеризира с васкулит на малки съдове, придружени от грануломи на Wegener и еозинофилни инфилтрати в Churg-Strauss.
При тип I бързо прогресиращ гломерулонефрит има линейни, глобални и дифузни отлагания на IgG (Фигура 3), обикновено придружени от по-малки количества линейни С3. Понякога има линейно оцветяване за IgM или IgA с по-малка интензивност. Много необичайно е наличието на IgA без IgG. Преобладаващият IgG е IgG1 подклас. Липсва линейно оцветяване на IgG в тубуларните мембрани в повече от 50% от случаите. Също така има линейно оцветяване в базовите мембрани на алвеоларните капиляри, но оцветяването е по-неравномерно, с по-малка интензивност, повече фон и по-трудно за интерпретация, отколкото с бъбречно оцветяване.
При тип III бързо прогресиращ гломерулонефрит няма натрупвания на имуноглобулини или комплементи. В някои случаи може да се идентифицира слабо имунооцветяване, но ако се открие силно оцветяване, трябва да мислим за тип I (ако има линейни отлагания) или за имунни комплекси (ако има грануларно оцветяване). Некротизиращите сегменти могат да имат неспецифично свързване на IgM и/или СЗ.

Библиография
https://emedicine.medscape.com/article/240457-overview
https://en.wikipedia.org/wiki/Rapidly_progressive_glomerulonephritis
http://www.kidneypathology.com/English_version/Crescentic_GN.html
http://www.pathologyoutlines.com/topic/kidneyrpgn.html
https://www.sciencedirect.com/topics/medicine-and-dentistry/rapidly-progressive-glomerulonephritis
Коментари към Бързо прогресиращ гломерулонефрит