Автозомно рецесивно поликистозно бъбречно заболяване
Автозомно рецесивно поликистозно бъбречно заболяване е рядко генетично заболяване. Характеризира се с кистозна дилатация на бъбречни събирателни канали, свързани с чернодробни аномалии в различна степен, включително билиарна дисгенезия и перипортална фиброза. За първи път е признато през 1902 година. През 1964 година Осатъндън и Потър класифицират автозомно рецесивно поликистозно бъбречно заболяване като кистично бъбречно заболяване тип 1. Автозомно рецесивно поликистозно бъбречно заболяване първоначално е описано като 4 отделни клинични единици въз основа на възрастта на представяне. Тази класификация вече не се счита за валидна поради голямата степен на припокриване между различните групи и широкия спектър от възможни презентации, независимо от възрастта.
Заболяването засяга мъже и жени на еднакъв брой. Честотата се оценява на около 1 на 20 000 индивида в общата популация. Тъй като някои хора могат да не диагностицират, е трудно да се определи истинската честота в общото население. Въпреки че повечето пациенти са диагностицирани вътреутробно или при раждането, леките случаи може да не станат видими до юношеството или зрелостта. Автозомно рецесивно поликистозно бъбречно заболяване може да засегне хора от всяка етническа група.
Автозомно рецесивно поликистозно бъбречно заболяване се причинява от мутации на PKHD1 гена, който се унаследява като автозомна рецесивна черта. Изследователите са установили, че повечето типични случаи на автозомно рецесивно поликистозно бъбречно заболяване са причинени от мутация към един единствен ген, по-специално на PKHD1 гена. Този ген се намира на дългото рамо на хромозома 6.
Установено е, че PKHD1 генът съдържа инструкции за създаване (кодиране) на протеин, известен като фиброцистин (или полидуктин). Ако пациентите имат две мутации, които не водят до генериране на протеин, резултатът обикновено е летален. Въпреки това, при мнозинството от пациентите поне едно копие на гена генерира някои функционални протеини и тези случаи обикновено са жизнеспособни. Точната роля и функция на този протеин в тялото не е известна. Протеинът укрепва теорията, че основният дефект при автозомно рецесивно поликистозно бъбречно заболяване е свързан с цилиарната дисфункция.
Всички бъбречни кисти се развиват от фокална епителна пролиферация някъде по протежението на нефрона. При автозомно рецесивно поликистозно бъбречно заболяване това се случва предимно в събирателния канал. Симетричната и периферната епителна пролиферация води до тубулно удължаване и фузиформна дилатация на събирателния канал. Анормалният епител също проявява необичайна промяна във функцията. Докато нормалният епител е по същество абсорбиращ (тоест резорбира течност от лумена на канала и го транспортира през епитела и в интерстициума на бъбреците), абнормалният пролиферативен епител парадоксално става секреторен. Флуидът, секретиран в лумена на ектатичния канал, е богат на епителни растежни фактори, които стимулират по-нататъшна епителна пролиферация.
Автозомно рецесивно поликистозно бъбречно заболяване се характеризира с неструктурна, двустранна, симетрична дилатация и удължаване на 10-90% от бъбречните събирателни канали, фокално отчитащи широка вариабилност на бъбречната дисфункция. Тъй като броят на участващите канали се увеличава, бъбреците се увеличават. При аутопсията обаче се бъбречната форма е запазена, тъй като аномалията е в събирателните канали, а кистите обикновено са малки (под 3 милиметра). При по-възрастни пациенти могат да се наблюдават кисти с големина до 1 сантиметър.
Макроскопското изследване на бъбреците при пациенти с автозомно рецесивно поликистозно бъбречно заболяване разкрива множество малки кистични пространства в капсулната повърхност. При срез на бъбреците се вижда, че кистичните структури са подкапсулни разширения на радиално ориентирани цилиндрични или фузиформени ектатични пространства с лоша кортикомедуларна диференциация, дължаща се на удължаване на продълговатите и разширени събирателни канали от медулата до кортекса.
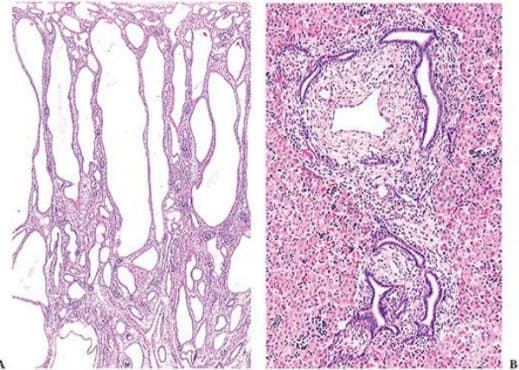
При хистологичен анализ, бъбрекът показва множество разширени и удължени тубулни структури, радиално ориентирани по отношение на бъбречния хилум (фигура 1). Електронната микроскопия показват, че обструкцията не е основната причина за дилатация. Кистите всъщност представляват дилатация и хиперплазия на интерстициалните части на клоните на уретерните пъпки, които образуват събирателните канали. Нефронната индукция е незасегната и броят на образуваните гломерули е нормален. На тъканни срезове, плътността на гломерулите изглежда намалява поради отделяне от разширени събирателни канали и от наличието на променливи степени на интерстициален оток и фиброза.
Всички пациенти с автозомно рецесивно поликистозно бъбречно заболяване имат вродена чернодробна фиброза, която може да има по-тежка клинична проява от бъбречното заболяване. Вродената чернодробна фиброза е резултат от малформация на развиващата се дуктуална плака. Констатациите от чернодробната биопсия показват разширени, фиброзни портални пътища и хиперпластични, разширени и дисгенетични жлъчни канали с нормални хепатоцити (фигура 2). Дуктулите могат да покажат истински кистични промени, а когато промените са макроскопични, автозомно рецесивно поликистозно бъбречно заболяване може да бъде неразличимо от болестта на Кароли.
Клиничните прояви на автозомно рецесивно поликистозно бъбречно заболяване варират в зависимост от броя на участващите събирателни канали, както и от степента на интерстициална фиброза. Фетуси с тежко увреждане на бъбречната функция и намалена продукция на урина са налице с олигохидрамнион, което може да доведе до белодробна хипоплазия. Повечето от тези деца умират от белодробни усложнения след раждането.
Бебетата с по-леки бъбречни прояви, които преживяват неонаталния период, все още могат да развият хронично бъбречно заболяване, което се проявява при различни възрасти в зависимост от степента на бъбречно засягане. Белодробната недостатъчност с респираторен дистрес, дължаща се на олигохидрамнион, е основна причина за заболеваемост и смъртност при новородени. При пациенти, които преживяват новородения период, бъбречната прогноза се е подобрила с течение на времето поради бъбречна трансплантация.
Библиография
https://en.wikipedia.org/wiki/Autosomal_recessive_polycystic_kidney_disease
https://rarediseases.org/rare-diseases/autosomal-recessive-polycystic-kidney-disease/
https://emedicine.medscape.com/article/983281-overview#a4
http://www.pathologyoutlines.com/topic/kidneytumorchildpkd.html
https://pubs.rsna.org/doi/full/10.1148/radiographics.20.3.g00ma20837
Коментари към Автозомно рецесивно поликистозно бъбречно заболяване