Вродени нарушения в синтеза на колаген
Вродените нарушения на колагеновата синтеза представляват една голяма група от състояния, проявяващи се клинично по различни начини. Най-известни са osteogenesis imperfecta, синдром на Ehlers-Danlos и синдром на Марфан. При тези три заболявания се наблюдава в различна степен нарушение на колагеновата синтеза.
Синдромът на Марфан е наследствено заболяване на съединителната тъкан, със сравнително високо разпространение и клинична вариабилност. Този автозомен доминантен синдром има плейотропни прояви, включващи предимно очните, сърдечно-съдовите и скелетните системи. Класически синдромът на Марфан се счита за състояние, причинено от дефицита на структурен протеин на извънклетъчна матрица, фибрилин-1. Проучвания на модели с мишки разширяват настоящото разбиране към патогенен модел, който включва нарушаване на сигнализацията на цитокин-трансформиращ растежен фактор бета (TGFβ). Пациенти, които имат клинични открития на заболяването, както и генетични варианти в гена на трансформиращ растежен фактор-бета рецептор-1 (TGFβR1) и гена на трансформиращ растежен фактор-бета рецептор-2 (TGFpR2), са означени като притежаващи синдром на Марфан тип 2.
Синдромът на Марфан е резултат от хетерозиготни мутации във фибрилин-1 ген, разположен на хромозома 15, която кодира гликопротеин фибрилин. Фибрилин е основен градивен елемент от микрофибрили, които съставляват структурните компоненти на суспензионния лигамент на очната леща и служат като субстрати за еластин в аортата и други съединителни тъкани. Аномалиите, включващи микрофибрилите, отслабват аортната стена. Прогресивната аортна дилатация и евентуална аортна дисекация се дължат на напрежението, причинено от импулсите на изтласкване на лявата камера. По подобен начин дефицитното отлагане на фибрилин води до понижена структурна цялост на зонулите на лещата, на връзките, на дихателните пътища на белия дроб и на гръбначния стълб.
Производството на анормални фибрилин-1 мономери от мутиралия ген нарушава мултимеризацията на фибрилин-1 и предотвратява образуването на микрофибрила. Този патогенен механизъм е наречен доминантно-отрицателен, тъй като анормалният фибрилин-1 нарушава образуването на микрофибрила (въпреки че други фибрилинови гени все още кодират нормален фибрилин). Доказателство за този механизъм е показано при проучвания на култивирани кожни фибробласти от пациенти със синдром на Марфан, които продуцират значително намалени и анормални микрофибрили.
Проучванията предполагат, че аномалиите в трасиращия път на трансформиращ растежен фактор-бета (TGFβ) може да представляват общ път за развитието на Марфан-фенотип. Този ген дефект в крайна сметка води до понижено и неправилно включване на фибрилин в матрицата на съединителната тъкан.
Различават се два типа на морфогенеза на заболяването:
- Колагенов тип - синтезират се колагенови молекули, които не могат да се свързват помежду си, в резултат на което стават неустойчиви.
- Еластинов тип - характеризира се с дефект в структурирането на еластиновите влакна.
Като се има предвид променливата експресивност на синдром на Марфан, нито един знак не е патогномичен. Диагнозата се прави на клинични основания въз основа на типични аномалии. Сърдечните, скелетните и очните системи обикновено са по-фокусирани върху диагностичните критерии. Въпреки това, в това състояние могат да бъдат засегнати и други тъкани, включително скелетните мускули, мастната тъкан, кожата, фасцията и дихателните пътища.
Заболяването се демонстрира с абнормно дълги крака и ръце, дълги и тънки пръсти на ръцете, поради значителната дължина на метакарпалните и фалангиалните кости. Наблюдава се дислокация на очната леща, поради слабост във фиксиращата й система. От страна на сърдечносъдовата система се открива регургитация на митралната клапа с прогресираща сърдечносъдова недостатъчност.
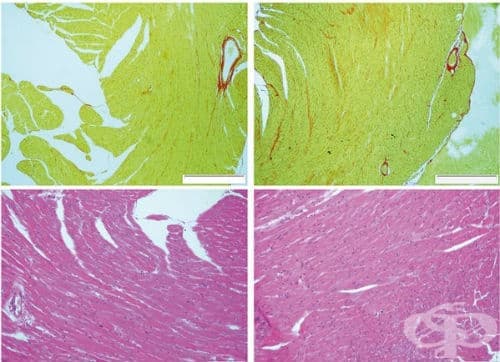
Макроскопски се откриват патологични промени в костния скелет, аортата и сърцето. В аортата се наблюдава развитие на медионекроза и аневризми на торакалната й част. Смъртта настъпва най-често при състояние на дисекация на аортната стена. В сърцето се открива увреждане на митралната клапа, която е със задебелени и уплътнени платна, което води до пролапс на митралната клапа към лявото предсърдие.
Микроскопски се наблюдава медионекроза в медията на аортата. В зоната на медионекрозата се открива деструкция на еластични ламели и гладкомускулни клетки и отлагане на гликозамингликани. В сърцето клапното платно се пропива от миксоматозна тъкан, богата на гликозамингликани.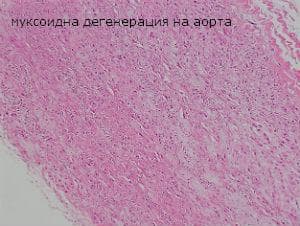
Морфологично, тъканта на скелетната мускулатура показва запазена архитектура, нормални ендо- и перимисиални компоненти и многоъгълни влакна с леки вариации в диаметъра. Умереният брой на миофибрилите показва множество фини вакуоли, придаващи ситов вид. Тези влакна присъстват между нормално появяващите се влакна, които дават мозаечен модел. Вакуолите са позитивни на червено О и отрицателни за PAS и кисела фосфатаза. Малки парчета тъкан на скелетната мускулатура, фиксирани в 3% глутаралдехид, показват изкривяване на филаментозния модел, наличие на липидни вакуоли и агрегати на митохондрии с различни размери с променени митохондриални кристали.
По отношение на мускулната патология се наблюдават значително повишени липидни капчици както по брой, така и по размер в мускулните влакна, особено при влакна тип 1. Ултраструктурното изследване често показва липидни капчици до митохондриите, които обикновено се увеличават. Наблюдавани са анормални митохондрии с различен размер с анормална кристална конфигурация. Цитоплазмични тела с плътно ядро и радиално разположени лептомери и филаменти също са демонстрирани в някои миофибрили.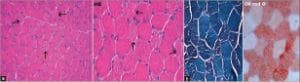
Библиография
http://emedicine.medscape.com/article/1258926-clinical#b4
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3548382/
https://en.wikipedia.org/wiki/Marfan_syndrome
https://medlineplus.gov/marfansyndrome.html
Коментари към Вродени нарушения в синтеза на колаген