Кумулативни муколипидози

Кумулативните муколипидози са група от заболявания, които са междинни, между мукополизахаридозите и липидозите. Муколипидозата е група от наследствени метаболитни нарушения, които засягат способността на организма да осъществява нормалното преместване на различни материали в клетките. В тъканите се отлага глюкозоаминогликани и GM1. Кумулативни муколипидози се различават по характера на ензимния дефект. Те се характеризират с амавротична идиотия, с поражение на нервната система, увреждане на вътрешните органи и скелета.
Когато първоначално са били посочени, муколипидозите получили своето име от сходството при представянето както на мукополизахаридози, така и на сфинголипидози. Биохимичното разбиране на тези условия променя начина, по който те се класифицират. Въпреки че четири състояния (I, II, III и IV) са били маркирани като муколипидози, тип I (сиалидоза) сега се класифицира като гликопротеиноза, а тип IV сега се класифицира като ганглиоидоза. Другите два вида кумулативни муколипидози са тясно свързани. Муколипидоза тип II и III са резултат от дефицит на ензима N-ацетилглюкозамин-1-фосфотрансфераза, който фосфорилира целеви въглехидратни остатъци върху N-свързани гликопротеини. Без това фосфорилиране, гликопротеините не са предназначени за лизозоми и те избягват извън клетката.
При разстройства на лизозомното съхранение дефицитът на специфичен лизозомен ензим прекъсва нормалния катаболитен път, водещ до клетъчно натрупване на субстрати, обикновено разградени от този ензим. Специфичността на тези натрупани материали за отделен ензимен дефект е учудващо в лизозомните разстройства и натрупването след това води до анормална клетъчна архитектура. Точно как промените в клетъчната структура, дължащи се на съхранение, се превръщат в неблагоприятни ефекти върху клетъчната функция остават загадъчни. Някои доказателства включват невраминидаза в регулирането на вътреклетъчния трафик на лизозомнияLAMP-1 мембранен протеин. LAMP-1 може да бъде инструмент за лизозомна екзоцитоза. Необходими са допълнителни проучвания, за да се обясни напълно ролята на невраминидазата в този процес.
Клиничният ход на кумулативни муколипидози зависи от свързаните с него ефекти на прогресивно съхранение в органовите системи, където тези субстрати са силно концентрирани. При сиалидоза дефицитът на лизозомната алфа-N-ацетилна нераминидаза предотвратява нормалното разграждане на гликопротеини, съдържащи остатъци на сиаловата киселина. Това води до вътрешноклетъчно съхранение на излишък на сиалилолигозахариди и е хистологично наблюдавано като анормална вакуолизация наразлични клетъчни типове. Докато костният мозък и циркулиращите лимфоцити са силно вакуолирани при сиалидоза тип II, тези открития очевидно отсъстват при заболяване от тип I. Системите на органите, които са най-вече включени в сиалидозата, включват централна нервна система, скелетната система и ретикулоендотелната система.
Муколипидозата тип I е рядко наследствено заболяване на лизозомното съхранение, което има клинични и хистологични данни, подобни на мукополизахаридозите и сфинголипидозите. В края на 60-те години на миналия век се съобщава за малък брой пациенти със скелетна дисплазия, психомоторно забавяне и нормална екскреция на мукополизахаридите в урината. Първоначално се класифицира като липомукополизахаридоза, това заболяване по-късно се класифицира в групата на подобни заболявания, известни като кумулативни муколипидози. При пациентите с муколипидоза тип 1 впоследствие се открива изолиран дефицит на алфа-N-ацетил невраминидаза (сиалидаза) в левкоцити и култивирани фибробласти и по този начин имат повишени количества сиалилолигозахарид в урината.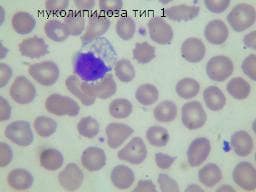
Муколипидоза тип I клинично се характеризира с псевдогаргоилизъм. Ензимният дефект се изразява в пълно отсъствие на А, В и С фракции на бета-галактозидаза. Количеството на GM1 в главния мозък се увеличава до 30 пъти над нормата. Макроскопски се наблюдават промени в скелета сходни с тези на гаргоилизма, хепато- и спленомегалията.
Микроскопски в ганглийните клетки се натрупва ПАС - положителна субстанция. В черния дроб, слезката, тимуса, костния мозък и лимфните възли, както и епителните клетки на бъбречните каналчета се откриват "пенести" клетки. В периферни лимфоцити, клетки на костен мозък, конюктивен епител, хепатоцити, тъканни фибробласти, проби от нервна биопсия, минетерни плексусови неврони и материал от мозъчна биопсия се наблюдава цитоплазмена вакуолизация в различна степен. Електронно микроскопски невроните и олигодендроглиоцитите показват натрупване в тях на осмиофилни телца с ламеларен и зърнист строеж. Ендоплазматичният ретикулум е с разширени цепки. В около ядреното пространство се виждат оптически плътни телца.
Муколипидоза тип II е наследствено разстройство на лизозомното съхранение. Първоначално е описано през 1967 година от Leroy и DeMars, когато съобщават за пациент с клинични и рентгенографски признаци, подобни на тези на синдрома на Хърлър (мукополизахаридоза тип 1), но с по-ранна поява на симптоми и без доказателства за мукополизахаридоза. Една уникална характеристика на това заболяване е наличието на фазовоплътни интрацитоплазмични включвания във фибробластите на пациентите. Тези клетки се наричат клетки на включване или I-клетки. По този начин заболяването се определя като I-клетъчна болест. Spranger и Wiedermann впоследствие класифицират тази болест като муколипидоза тип II, тъй като тя има клинични характеристики, които включват мукополизахаридози и сфинголипидози.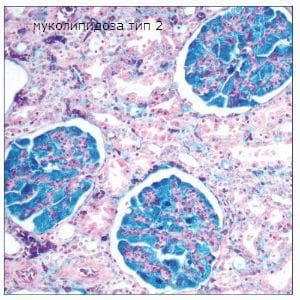
Муколипидоза тип II се наблюдава в ранната детска възраст и клинично се демонстрира с прогресивно нарушение функциите на централната нервна система. Ензимният дефект засяга В и С фракциите на бета-галактозидазата. Ранни ензимологични изследвания показват, че култивираните фибробласти от пациенти с I-клетъчно заболяване са с недостатъци в много лизозомни ензими. Освен това е установено, че тези ензими присъстват в излишък в тъканна културелна среда и в извънклетъчни течности, като серум и урина. Впоследствие са открити фибробласти на I-клетъчно заболяване, които са в състояние да интернализират и използват лизозомни ензими, продуцирани от нормални клетки, докато нормалните или други лизозомни заболявания фибробласти не са в състояние да интернализират лизозомните ензими, секретирани от фибробластите на I-клетъчната болест.
Както при много от болестите на лизозомното съхранение, функционалният дефицит на лизозомните ензими води до анормална клетъчна архитектура. При I-клетъчно заболяване характерната находка е анормална вакуолизация или включвания, които се появяват в цитоплазмата. Те се наблюдават в клетки с мезенхимен произход, особено фибробласти. Най-силно засегнатата система е скелетната система, при която трабекулацията на костните и хрущялните структури е ненормална. Мускулната тъкан, включително сърдечния мускул, е сравнително пощадена. Обаче, значителна вакуолизация присъства в клетките на съединителната тъкан на сърдечните клапи. Това води до удебеляване на клапите, което води до клинично значимо клапно заболяване. Други места на анормална вакуолизация на клетките включват бъбречните гломерулни подоцити и във фибробластите в перипорталните пространства на черния дроб. Хепатоцитите и Купферовите клетки не са засегнати.
Уникална находка при хистологията на I-клетъчно заболяване е наличието на множество интрацитоплазмични включвания в клетки от мезенхимален произход, които се наблюдават при електронна микроскопия. Тези включвания са мембранно свързани вакуоли, пълни с фибрилогрануларен материал. Съдържанието на тези вакуоли не е добре характеризирано. Обаче, те съдържат различни липиди, мукополизахариди и олигозахариди.
Муколипидоза тип III е лизозомно нарушение, характеризиращо се с прогресивно забавяне на темпа на растеж от ранна детска възраст, скованост и болка в ставите, постепенно закръгляване на черти на лицето, умерено забавяне на развитието и леко умствено увреждане при повечето пациенти. Муколипидозата тип III е резултат от дефицит на ензима N-ацетилглюкозамин-1- фосфотрансфераза. Недостатъчният ензим трябва да маркира други ензими (активаторни протеини), така че те да могат да инициират определени метаболитни процеси в клетката. Тъй като протеините на активатора не са правилно маркирани, те избягват извън клетката и следователно не могат да извършват обичайната си работа за разграждане на веществата вътре в клетките.
При електронна микроскопия мезенхимните клетки във всяка тъкан разкриват голям брой цитоплазмени вакуоли, включващи набъбнали лизозоми, свързани с единична мембрана. Съдържанието е плеоморфно, но не и плътно. Това явление е специфично за муколипидоза тип III и не се наблюдава при всяко лизозомно запаметяващо заболяване. Активността на лизозомните ензими е силно редуцирана в I-клетките, но значително се увеличава в съответната културална среда. Цитологичните и ензимните находки в клетъчната култура не могат да разграничат от муколипидоза тип II.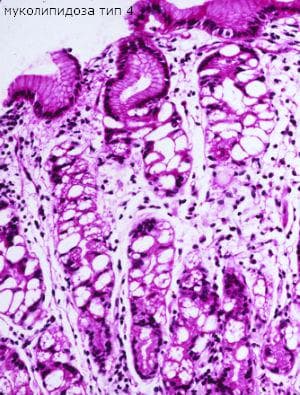
Муколипидозата тип IV е автозомно рецесивно разстройство на лизозомното съхранение. Хората с нарушение имат много симптоми, включително забавено психомоторно развитие и различни очни аберации. Разстройството се причинява от мутации в MCOLN1 гена, който кодира неселективен катионен канал, муколипин 1. Тези мутации нарушават клетъчните функции и водят доневроразвиващо разстройство чрез неизвестен механизъм. Предполага се, че муколипинът се локализира в ендозоми. Важно свойство на муколипин 1 е, че намаляването на рН (подкисляването) води до деактивиране на протеина, вероятно чрез сглобяване на дефекта. Има най-малко 29 известни мутации в MCOLN1, разположени в целия ген. Много от известните мутации не водят до експресия на муколипин 1. По-слабите мутации продуцират дисфункционална форма на катионния канал. Мутациите, които променят само С-края на протеина, също водят до лек фенотип на разстройството, обикновено съхраняващ мозъка. При муколипидоза тип IV в засегнатите клетки се натрупват автофлуоресцентни вакуоли, считани за анормални лизозоми.
В миналото идентифицирането на анормални структури на ламеларна мембрана и аморфни цитоплазмични включвания в различни клетъчни видове при кожна биопсия се използва за потвърждаване на диагнозата муколипидоза IV. Впоследствие се използва диагностика на типична вакуолация чрез PAS оцветяване на конюнктивални клетки, получени с тампон.
Заглавно изображение: magnific.com
Библиография
https://www.ncbi.nlm.nih.gov/books/NBK1214/
https://en.wikipedia.org/wiki/Mucolipidosis
http://emedicine.medscape.com/article/945460-overview#a5
http://emedicine.medscape.com/article/945460-workup#c7
http://www.ldnz.org.nz/lysosomal_diseases/list_of_lysosomal_disorders/mucolipidosis_type_iii
Коментари към Кумулативни муколипидози