Имунодефицитни синдроми, свързани с комбиниран дефект на клетъчния и хуморален имунитет
Тежката комбинирана имунна недостатъчност е група от редки вродени синдроми с малко или никакви имунни отговори. Това води до често повтарящи се инфекции с бактерии, гъбички и вируси. Инфекциите, които са незначителни за повечето хора, могат да бъдат животозастрашаващи при хора с имунодефицитни синдроми, свързани с комбиниран дефект на клетъчния и хуморален имунитет. Имунната система включва специализирани бели кръвни клетки, които работят заедно за борба с бактериите, гъбите и вирусите. Тези бели кръвни клетки включват Т-лимфоцити (Т-клетки), които са централни медиатори на имунния отговор, а също и директно атакуват вируси. В-лимфоцитите (В-клетките) произвеждат антитела, които се прикрепят към чуждите тела и ги маркират, за да бъдат фагоцитирани. NK-клетките са специализирани да спомагат и за борбата с вирусите.
Пациентите с имунодефицитни синдроми, свързани с комбиниран дефект на клетъчния и хуморален имунитет имат генетичен дефект, който засяга Т-клетките и най-малко един друг тип имунни клетки. Има няколко вида имунодефицитни синдроми, всеки от които е причинен от друг генетичен (наследствен) дефект.
В рубриката имунодефицитни синдроми, свързани с комбиниран дефект на клетъчния и хуморален имунитет ще бъде разгледан по- подробно синдром на Гланцман и Риникер.
Тежката комбинирана имунна недостатъчност, наричана още синдром на Гланцман и Риникер, е животозастрашаващ синдром на повтарящи се инфекции, диария, дерматит. Той е прототип на първичните имунодефицитни заболявания и е причинен от множество молекулни дефекти, които водят до тежко увреждане в броя и функцията на Т-клетките, В-клетките и понякога NK-клетките. Без намеса синдромът обикновено води до тежка инфекция и смърт при деца до 2-годишна възраст.
Тежката комбинирана имунна недостатъчност е резултат от мутации в някой от повече от 15 познати гени. Тези молекулни дефекти пречат на развитието и функцията на лимфоцитите, блокирайки диференциацията и пролиферацията на Т-клетките и в някои видове В-клетки и NK-клетки. Производството на антитела е силно нарушено, дори когато са налице зрели В-клетки, поради липса на Т- клетъчна помощ. NK-клетките, компонент на вродения имунитет, са променливи. Последователността и други техники могат да разкрият действителните генетични дефекти при тези пациенти.
Патогенезата на тежката комбинирана имунна недостатъчност може допълнително да се раздели на следните 5 механизма въз основа етапите на увреждане на лимфопоезата:
- Дефектен лимфокинен сигнал- може да се появи ранен блок в рамките на пътя на диференциацията на Т-клетките. Най-често срещаната форма на синдрома, възникваща при 40-60% от пациентите, е XL-формата, която възниква от дефекти в общата верига на рецепторите на интерлевкин. Този молекулен дефект води до отсъствие на Т-клетъчна и NK-клетъчна зрялост.
- Апоптоза, вследствие на натрупване на токсични метаболити- анормален пуринов метаболизъм може да бъде включен в патогенезата на синдрома на Гланцман и Риникер. Това води до натрупване на междинни продукти (например аденозин дифосфат, гуанозин трифосфат), което води до токсичност на лимфоцитите, особено при незрели тимусни лимфоцити.
- Дефектна клетъчна сигнализация - CD45, тирозин фосфатаза, намираща се в клетъчните мембрани на хематопоетични клетки, е от съществено значение за регулиране на предаването на сигналите на клетъчната повърхност в В-клетките и Т-клетките.
- Дефектно пренареждане на TCR и Ig гени- може да възникне анормално пренареждане на TCR и Ig гени. Както съзряването на В- клетките, така и узряването на Т-клетките включват процес на рекомбинация, в който се съберат различни комбинации от VDJ гени за създаване на уникални и специфични антигенни рецептори. Няколко рекомбинации играят важни роли в този процес.
- Тимусна дисгенеза - води до липса на Т-клетки

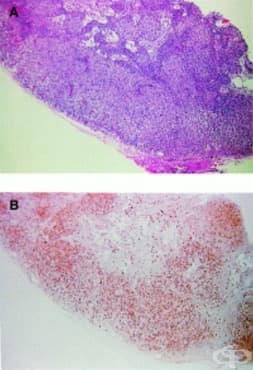
При класическия синдром на Гланцман и Риникер, спадащ към имунодефицитни синдроми, свързани с комбиниран дефект на клетъчния и хуморален имунитет, тимусът е хипопластичен. Кожата и червата могат да покажат инфилтрация с хистиоцити, еозинофили или активирани дисфункционални Т-клетки. Епидермисът може да има огнища на хиперкератоза с паракератоза или неправилна акантоза със спонгиоза и екзоцитоза. Папиларния дермис има оток и дифузен периваскуларен инфилтрат с някои еозинофили.
Слезката и периферните лимфни възли са характерно атрофични, но понякога могат да бъдат хиперпластични, с хистиоцити и еозинофили. Слезката е изчерпана от лимфоцити. Въпреки че биопсията на лимфните възли не е необходима за диагностициране, находките могат да показват липса на Т- и В-клетки и липса на зародишни центрове. Тонзилите и аденоидите са недоразвити или липсват.
Библиография
Color atlas of pathology, Section Immune pathology
https://rarediseases.org/rare-diseases/severe-combined-immunodeficiency/
https://primaryimmune.org/about-primary-immunodeficiencies/specific-disease-types/severe-combined-immune-deficiency-and- combined-immune-deficiency
https://emedicine.medscape.com/article/888072-overview
Коментари към Имунодефицитни синдроми, свързани с комбиниран дефект на клетъчния и хуморален имунитет