Автозомно доминантно поликистозно бъбречно заболяване
Автозомно доминантно поликистозно бъбречно заболяване е мултисистемно и прогресивно разстройство, характеризиращо се с образуването и разширяването на кисти в бъбреците и други органи (например черен дроб, панкреас, слезка). Клиничните признаци обикновено започват през третото до четвъртото десетилетие от живота, но кистите може да се открият в детска възраст и по време на вътреутробното развитие на плода.
Автозомно доминантно поликистозно бъбречно заболяване е най-разпространеното, потенциално летално моногенно човешко заболяване. То се свързва с голяма междуфамилна и вътрешнофамилна вариабилност, което може да бъде обяснено до голяма степен от неговата генетична хетерогенност и модифициращи гени. Също така е най-честата от наследените муковисцидозни заболявания - група от заболявания със сходна, но различна патогенеза, характеризираща се с развитие на бъбречни кисти и различни извън бъбречни прояви. Над 50% от пациентите с автозомно доминантно поликистозно бъбречно заболяване накрая развиват краен стадий на бъбречно заболяване и изискват диализа или бъбречна трансплантация. Заболяването се оценява, че засяга най-малко 1 на всеки 1000 души по света, което прави това заболяване най-разпространеното наследено бъбречно заболяване с диагнозно разпространение 1:2000 и честота 1:3000-1:8000 в световен мащаб.
Автозомно доминантно поликистозно бъбречно заболяване е наследствено заболяване. Начинът на наследяване е автозомно доминантен. Тъй като разстройството се среща еднакво при мъже и жени, всяко потомство има 50% шанс да наследи отговорната мутация и следователно болестта. Тя е генетично хетерогенно състояние, което включва най-малко 2 гена. Около 86% от случаите на автозомно доминантно поликистозно бъбречно заболяване са причинени от мутация на PKD1 гена на хромозома 16. Повечето от останалите се причиняват от мутации на PKD2 гена на хромозома 4. Тези две заболявания са фенотипно почти еднакви, различаващи се само по-високата възраст на диагностициране с PKD2 и по-бавното му прогресиране до крайна фаза на бъбречно заболяване.
При много пациенти с това заболяване бъбречната дисфункция не е клинично видима до четиридесетата или петдесетата година от живота. Има обаче все повече доказателства, които предполагат, че образуването на бъбречни кисти започва в утробата. Кистите първоначално се образуват като малки дилатации в бъбречните тубули, които след това се разширяват, за да образуват кухини, пълни с течност, с различни размери. Факторите, за които се предполага, че водят до кистогенеза, включват мутация на зародишна линия в един от генните алели, кодиращи полицистин. Това се нарича "соматичен втори удар", който води до загуба на нормалния алел и "трети удар", който може да бъде всичко, което предизвиква клетъчна пролиферация, което води до разширяване на тубулите. При прогресирането на заболяването продължителното разширяване на тубулите чрез увеличена клетъчна пролиферация, секреция на течности и отделяне от родителските тубули води до образуването на кисти.
Автозомно доминантно поликистозно бъбречно заболяване, заедно с много други заболявания, които се срещат с бъбречни кисти, могат да бъдат класифицирани в семейство от заболявания, известни като цилиопатии. Епителните клетки на бъбречните тубули, включващи всички сегменти на нефрона и събирателните канали (с изключение на интеркалираните клетки), показват наличието на единичен първичен апикален цилиум. Полицистин-1, протеинът, кодиран от PKD1 гена, присъства на тези ресни и се смята, че усеща потока с големите си извънклетъчни домени, активирайки калциевите канали, свързани с полицистин-2, продукт на ген PKD2.
Пролиферацията на епителните клетки и секрецията на течности, които водят до кистогенеза, са две отличителни черти в автозомно доминантно поликистозно бъбречно заболяване. По време на ранните стадии на кистогенеза кистите се прикрепват към техните родителски бъбречни тубули и производно на гломеруларния филтрат навлиза в кистите. След като тези кисти се разширят до диаметър приблизително 2 милиметра, кистата се затваря от родителския тубул и след това течността може да навлезе в кистите само чрез трансепителна секреция, която на свой ред се препоръчва да се увеличи поради вторичните ефекти от повишените вътреклетъчни концентрации на циклични аденозин монофосфат.
Клинично, коварното увеличаване на броя и размера на бъбречните кисти се трансформира като прогресивно нарастване на обема на бъбреците. Бъбречните кисти са свързани с прекомерна ангиогенеза, проявена от крехки съдове, простиращи се през стените им. Когато бъдат травмирани, тези съдове могат да изтекат кръвта в кистата, което я кара да се разраства бързо, което води до мъчителна болка. Ако кървенето продължава, тогава кистата може да се разпадне в събирателната система, причинявайки груба хематурия.
При автозомно доминантно поликистозно бъбречно заболяване и двата бъбрека са разширени, понякога до впечатляващ размер (до 25 сантиметра и 4 килограма, всеки), компресиращи коремните органи. Външната повърхност на бъбрека се деформира. На срез, кората и медулата представляват множество сферични кисти, с диаметър между 0,5 и 5 сантиметра, съдържащи серозна, хеморагична или желатинова течност. Между кистите, вливащият се паренхим е намален, атрофичен чрез компресия. Въпреки това, микроскопски този паренхим е представен от функционални нефрони. На фигура 1 е показано сравнение между здрав бъбрек, с нормални размери, и бъбрек на пациент с автозомно доминантно поликистозно бъбречно заболяване.
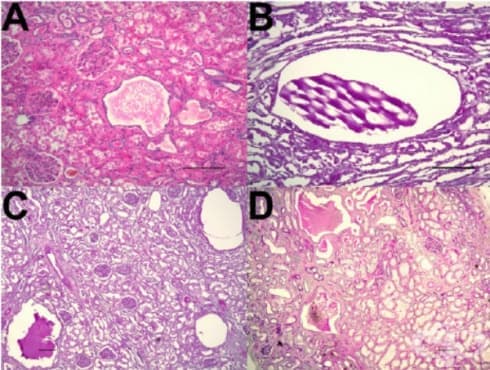
Микроскопски кистите са (разширени тубули и колекторни канали) облицовани с кубидален или сплескан епител, съдържат еозинофилна течност. Паренхимът между кистите е представен от няколко атрофични/компресирани, но все още функционални нефрони (гломерули и тубули, фигура 2), с интерстициална фиброза и хронично възпаление. Кистите са така гъсто разположени, че създават впечатление за пълно изконсумиране на паренхима, но микроскопски между тях се откриват единични запазени нефрони. Руптурата на някои кисти и изливът на съдържимото им поддържат хроничен възпалителен процес в интерстициума. Макар и рядко, от стената им може да се развие бъбречно клетъчен карцином.
Библиография
https://en.wikipedia.org/wiki/Autosomal_dominant_polycystic_kidney_disease
https://emedicine.medscape.com/article/244907-overview#a4
http://www.pathologyatlas.ro/kidney-polycystic-disease.php
https://academic.oup.com/qjmed/article/100/1/1/2258660
http://www.pathologyoutlines.com/topic/kidneytumoradultpkd.html
Коментари към Автозомно доминантно поликистозно бъбречно заболяване